L’angiosarcoma cutaneo ha una delle peggiori prognosi tra tutti i tumori cutanei. È molto aggressivo e ha alti tassi di recidiva locale. Il tasso di sopravvivenza a 5 anni è tra il 12% e il 34% secondo la maggior parte degli studi,1,2 ma può raggiungere il 62%.3 A differenza di altri sarcomi, il grado di differenziazione non è stato associato alla prognosi nell’angiosarcoma cutaneo.4
La forma classica dell’angiosarcoma cutaneo è una lesione edematosa non ben definita simile a un livido con una presentazione clinica ampiamente indolente nelle sue prime fasi. Si verifica sul viso o sul cuoio capelluto di pazienti anziani (angiosarcoma di Wilson-Jones) e rappresenta circa il 50% di tutti gli angiosarcomi cutanei primari.5-8 Altre due forme tipiche di angiosarcoma sono la sindrome di Stewart-Treves, che si sviluppa in aree di linfedema di lunga data ed è particolarmente comune nelle donne che hanno subito una mastectomia radicale,9-11 e l’angiosarcoma post-radiazione, che si sviluppa in aree di pelle irradiata, in particolare nella regione pettorale di donne con una storia di cancro al seno trattato con radioterapia.12-L’aspetto istopatologico dell’angiosarcoma cutaneo varia da forme relativamente differenziate con spazi vascolari riconoscibili coperti da cellule endoteliali prominenti con qualche atipia e un modello infiltrativo che seziona i fasci di collagene a forme più solide altamente indifferenziate composte da cellule fusate o epitelioidi con atipia e pleomorfismo considerevolmente maggiori e un maggior numero di mitosi. Gli spazi vascolari sono rari e i tumori possono a volte imitare il carcinoma.
Il trattamento principale per l’angiosarcoma cutaneo – e l’unico potenzialmente curativo se si ottengono margini liberi da malattia – è l’escissione chirurgica con ampi margini seguita da radioterapia locale e persino, secondo alcuni autori, dalla radiazione dei linfonodi regionali.6 Nella maggior parte dei casi, tuttavia, non è facile ottenere margini liberi da malattia a causa della diffusione subclinica estesa. Inoltre, questi tumori sono spesso multifocali. La chemioterapia ha un ruolo puramente palliativo nella gestione dell’angiosarcoma cutaneo.
Anche se l’angiosarcoma cutaneo è raro (rappresenta meno dell’1% di tutti i sarcomi), la maggior parte dei casi di angiosarcoma ha origine nella pelle. A causa della loro bassa prevalenza, gli angiosarcomi cutanei sono inclusi in serie di angiosarcomi viscerali o ossei, che hanno una prognosi ancora più infausta.1 Ci sono quindi poche grandi serie di angiosarcomi cutanei in letteratura a causa della carenza di casi uniformi a lungo termine.2,4,5,7,16 La gestione dell’angiosarcoma è inoltre spesso scoraggiante, soprattutto nei casi di malattia avanzata, che hanno una prognosi molto scarsa nonostante l’uso di trattamenti aggressivi fin dall’inizio. Motivati quindi dalle difficoltà associate alla gestione dell’angiosarcoma cutaneo e dalla poca letteratura disponibile, abbiamo studiato tutti i casi di angiosarcoma cutaneo trattati presso l’Instituto Valenciano de Oncología (IVO), a Valencia, Spagna, con l’obiettivo di identificare i fattori clinici, istologici e legati al trattamento eventualmente associati alla prognosi. Per fare questo, abbiamo esaminato le cartelle cliniche e i risultati clinici alla ricerca di dati esplorativi che potessero servire da guida per una diagnosi precoce, dato che i pazienti con malattia precoce e tumori piccoli hanno una probabilità di sopravvivenza notevolmente migliore.
Materiali e metodi
Abbiamo condotto uno studio osservazionale retrospettivo di tutti i casi di angiosarcoma cutaneo trattati all’IVO tra gennaio 2000 e dicembre 2015. Tutte le informazioni raccolte sono state estratte dalle cartelle cliniche dei pazienti, dall’archivio delle biopsie del dipartimento di patologia e dall’archivio fotografico del nostro dipartimento. Dei 20 casi inizialmente identificati, 4 hanno dovuto essere esclusi: 1 perché non c’era follow-up, un altro perché non c’era materiale sufficiente per determinare se il tumore era un emangioendotelioma o un angiosarcoma, e 2 perché i tumori non erano angiosarcomi primari. Questi 2 tumori erano stati inizialmente etichettati come angiosarcoma cutaneo perché tutti i vetrini istologici mostravano un coinvolgimento cutaneo del seno. Tuttavia, rivedendo i blocchi, abbiamo scoperto che il coinvolgimento cutaneo era secondario in entrambi i casi, e che il tumore primario era situato nel parenchima mammario da dove si estendeva fino alla pelle sovrastante.
I criteri di inclusione per lo studio erano risultati clinici suggestivi di angiosarcoma cutaneo e la conferma istologica della diagnosi con colorazione ematossilina-eosina dei campioni bioptici, supportata da studi immunoistochimici, che nella maggior parte dei casi hanno coinvolto la colorazione CD31, CD34, D240 e Ki-67.
Le seguenti variabili sono state studiate per ogni paziente: età, sesso, localizzazione e dimensioni del tumore, tipo di angiosarcoma (primario, post-radiazione, associato a linfedema), trattamento (chirurgia, radioterapia, chemioterapia), recidiva, metastasi, sopravvivenza e morte. Per i tumori post-radiazione e associati al linfedema, abbiamo anche registrato il tipo di tumore precedente e il numero di anni trascorsi dalla radioterapia o dal linfedema. Le variabili istologiche analizzate erano lo stato dei margini, il modello istopatologico (vasoformativo, solido o misto), il tipo di cellula predominante (epitelioide o fusiforme), la presenza di necrosi (sì, no), il livello di invasione (epidermide, derma, ipoderma, muscolo, osso), la reazione linfocitica, il modello infiltrativo e il numero di mitosi per 10 campi.
Risultati
Sono stati inclusi nello studio sedici casi di angiosarcoma cutaneo. Essi corrispondevano a 11 donne e 5 uomini di età compresa tra 35 e 83 anni (media, 67 anni; mediana, 71 anni). Dieci dei casi erano angiosarcomi post-radiazione (10 casi), 5 erano angiosarcomi di Wilson-Jones, e solo 1 era angiosarcoma associato a linfedema. La localizzazione più comune era il tronco (10 casi), seguito dalla testa e dal collo (5 casi). Le estremità superiori sono state coinvolte solo in 1 caso. La dimensione più piccola del tumore era di 1 cm e la più grande di 50 cm (media, 10 cm; mediana, 6,5 cm).
Sette dei pazienti avevano una storia di cancro (cancro al seno in 10 casi e seminoma in 1). Con l’eccezione di 1 caso di carcinoma lobulare invasivo, tutti i tumori al seno erano carcinomi duttali invasivi.
Il tempo medio tra la radioterapia e lo sviluppo dell’angiosarcoma nei 10 casi di angiosarcoma postradiazione era di 8,2 anni. Solo 1 dei casi è apparso entro 5 anni dalla radioterapia; il resto è apparso almeno 5 anni dopo.
Quattordici casi sono stati trattati chirurgicamente e la radioterapia adiuvante è stata utilizzata in 4 di questi. Otto pazienti hanno ricevuto la chemioterapia e questo è stato il primo e unico trattamento in 2 pazienti.
Doxorubicina e taxolo sono stati utilizzati ciascuno in 4 casi, ifosfamide è stato utilizzato in 3 casi, e paclitaxel e dacarbazina sono stati utilizzati in 1 caso ciascuno. La risposta alla chemioterapia è stata scarsa, e anche se quasi tutti i pazienti hanno mostrato una risposta parziale, la malattia è progredita in tutti i casi e i pazienti sono morti durante il follow-up (8/8).
Cinque pazienti avevano metastasi a distanza, che hanno coinvolto più siti nella maggior parte dei casi. I siti più comuni erano il polmone e il fegato.
Dieci dei 16 pazienti sono morti di angiosarcoma durante il follow-up. Gli altri 6 pazienti sono attualmente liberi dalla malattia. La durata media del follow-up era di 42,5 mesi (mediana, 26 mesi; range, 7-188 mesi).
Istologicamente, 8 casi avevano un modello di crescita solido, 4 avevano un modello vasoformativo, e 4 avevano un modello misto. Il tipo cellulare predominante era epitelioide in 14 casi e fuso in soli 2. La necrosi è stata osservata in 6 tumori e il modello infiltrativo era sottocutaneo nella maggior parte dei casi (n=10). Quattro casi erano confinati al derma e solo 2 interessavano i piani muscolari. I margini chirurgici non erano valutabili in 3 casi. Dei casi rimanenti, 8 avevano margini negativi e 5 avevano margini positivi. La reazione linfocitaria era lieve o moderata in 10 casi, intensa in 2 e inesistente in 4. Nei 14 casi con una reazione linfocitaria, l’infiltrato era peritumorale in 2 casi, intratumorale in 8 e misto in 2. C’era una media di 15 mitosi per 10 campi (range, 0-37 mitosi).
I risultati clinici e patologici più rilevanti sono riassunti nella tabella 1. I risultati del confronto tra sopravvissuti e non sopravvissuti sono riassunti nella Tabella 2.
Selezione dei risultati clinici e patologici per i 16 angiosarcomi cutanei.a
| Paziente | Età, y | Sesso | Tipo | Localizzazione | Dimensione, cm | Tempo dalla Rx, mo | Dose, Gy | Tumore precedente | Tipo di cancro al seno | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | F | PR | seno sinistro | 18 | 60 | 46 | Seno | IDC | |
| 2 | 71 | F | ST | Braccio sinistro | 12 | – | – | Seno | IDC | |
| 3 | 51 | F | PR | Regione sottomammaria destra | 1 | 57 | 50 | Petto | IDC | |
| 4 | 76 | F | WJ | Testa e collo | 3 | – | – | No | . | |
| 5 | 77 | F | PR | seno destro | 1 | 94 | 46 | Seno | IDC | |
| 6 | 71 | F | PR | Seno sinistro | 50 | 171 | 48 | Seno | IDC | |
| 7 | 48 | M | PR | Parete addominale | 2 | 96 | 26 | Seminoma | – | |
| 8 | 55 | F | PR | seno sinistro | 10 | 88 | 46 | Seno | ILC | |
| 9 | 69 | F | PR | Seno sinistro | 8 | 143 | 46 | Seno | IDC | |
| 10 | 76 | M | WJ | Guancia destra | 6 | – | – | No | – | |
| 11 | 35 | F | PR | Seno destro | 12 | 66 | 50 | Seno | IDC | |
| 12 | 57 | F | PR | Seno destro | 8 | 108 | 50 | Seno | IDC | |
| 13 | 68 | M | WJ | Capo e collo | 2 | – | – | – | No | – |
| 14 | 80 | M | WJ | Testa e collo | 15 | – | – | No | – | |
| 15 | 79 | M | WJ | Capo e collo | 2 | – | – | No | – | |
| 16 | 83 | F | PR | Seno sinistro | 3 | 110 | 50 | Seno | IDC | |
| X=67.1 | 11W, 5M | 10 RI, 5 WJ, 1 ST | 9 seno, 5 testa e collo, 1 addome, 1 arto superiore | X=10 | X=100.3 | X=45.8 | 10 seno, 1 seminoma |
10 tumori del seno: 9 IDC, 1 ILC |
| Paziente | Chirurgia | Margine, cm | Trattamento | Morte | Schema HP | Tipo di cellule | Necrosi | DoI | Miti/mm2 | Sopravvivenza, mo |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Sì | 0.2 | 13 | Sì | 1 | E | No | 3 | 7 | 24 |
| 2 | No | ns | 3 | Sì | 2 | E | No | 3 | 37 | 8 |
| 3 | Sì | 3.5 | 1 | No | 1 | E | No | 3 | 2 | 29 |
| 4 | Sì | 2 | 123 | Sì | 2 | E | No | 3 | 28 | 26 |
| 5 | Sì | ns | 1 | Sì | 2 | E | No | 2 | 14 | 8 |
| 6 | No | ns | 3 | Sì | 1 | E | No | 2 | 0 | 19 |
| 7 | Sì | ns | 1 | No | 3 | E | No | 3 | 6 | 187 |
| 8 | Sì | ns | 13 | Sì | 2 | E | Sì | 4 | 6 | 28 |
| 9 | Sì | ns | 13 | Sì | 3 | E | Sì | 3 | 22 | 24 |
| 10 | Sì | ns | 12 | Sì | 2 | E | Sì | 3 | 16 | 7 |
| 11 | Sì | 0.5 | 1 | No | 2 | SC | No | 3 | 18 | 76 |
| 12 | Sì | 0 | 13 | Sì | 3 | E | Sì | 3 | 36 | 26 |
| 13 | Sì | 2 | 12 | No | 3 | E | No | 3 | 5 | 95 |
| 14 | Sì | 2 | 123 | Sì | 2 | E | Sì | 4 | 17 | 24 |
| 15 | Sì | 2 | 1 | No | 2 | E | Sì | 2 | 6 | 53 |
| 16 | Sì | 3 | 1 | No | 1 | SC | No | 2 | 23 | 51 |
| 14 sì, 2 no | 1.68 | 14: Qx 4: Rt 8: Qt |
10 sì 6 no |
4 vasof 8 solido 4 misto |
14 E, 2 SC | 6 sì 10 no |
4 derma 10 ipoderma 2 muscolo |
X=15 | X=42.8 |
I pazienti che sono morti sono indicati in grassetto.
Trattamento SA, trattamento dell’angiosarcoma (1, chirurgia; 2, radioterapia; 3, chemioterapia); DoI, profondità di invasione (2, derma; 3, ipoderma; 4, muscolo); E, epitelioide; HP, istopatologico (1, vasoformativo; 2, solido; 3, misto); IDC, carcinoma duttale invasivo; ILC, carcinoma lobulare invasivo; M, uomo; ns, non specificato; PR, angiosarcoma post-radiazione; Rx, radiazione; SC, cellula fusata; ST, angiosarcoma Stewart-Treves; W, donna; WJ, angiosarcoma Wilson-Jones: X, media.
Confronto delle variabili tra sopravvissuti e pazienti morti per angiosarcoma cutaneo.
| Variabile | Vivissuto (n=6) | Morto (n=10) | |
|---|---|---|---|
| Età, media, y | 61 | 71 | |
| Donne | 3 | 8 | |
| Uomini | 3 | 2 | |
| Postradiazione | 4 | 6 | |
| Idiopatico | 2 | 3 | |
| Linfedema-associato | 0 | 1 | |
| Tronco | 4 | 6 | |
| Testa e collo | 2 | 3 | |
| Arti superiori | 0 | 1 | |
| Taglia, cm | 3.6 | 13.1 | |
| Tempo dalla radioterapia, mo | 82.25 | 110.6 | |
| Dose di radiazione, Gy | 44 | 47 | |
| Seno | 3 | 7 | |
| Seminoma | 1 | ||
| Chirurgia | 6 | 8 | |
| Radioterapia | 1 | 3 | |
| Chemioterapia | 0 | 8 | |
| Vasoformativo | 2 | 2 | |
| Solido | 2 | 6 | |
| Misto | 2 | 2 | 2 |
| Necrosi | 1 | 5 | |
| Derma | 2 | 2 | |
| Sottocutaneo | 4 | 6 | |
| Muscolo | 0 | 2 | |
| Mitosi | 10 | 18.3 | |
| Sopravvivenza, mo | 81.8 | 19.4 | |
Discussione
L’angiosarcoma cutaneo è un tumore molto raro, evidenziato dal fatto che abbiamo potuto raccogliere dati solo su 16 tumori diagnosticati in un periodo di 14 anni in un ospedale oncologico. Nel complesso, l’angiosarcoma cutaneo è un po’ più comune negli uomini anziani. Questo perché la forma più comune di angiosarcoma cutaneo nella popolazione generale è l’angiosarcoma primario della testa e del collo (noto anche come angiosarcoma idiopatico o di Wilson-Jones) (Fig. 1), che tende a colpire uomini anziani.5,7,17 L’angiosarcoma post-radiazione (Fig. 2) è ora la seconda forma più comune di angiosarcoma cutaneo a causa dell’aumento dell’uso della radioterapia rispetto alla mastectomia radicale per trattare il cancro al seno.2,13,18 Questo cambiamento ha anche portato ad una riduzione della frequenza dell’angiosarcoma associato al linfedema, che è attualmente la forma meno comune di questo tumore. La prevalenza delle diverse forme di angiosarcoma cutaneo nella nostra serie non coincide con quanto riportato in letteratura, in quanto il nostro ospedale tratta un gran numero di donne con cancro al seno, spiegando perché la forma più comune di angiosarcoma cutaneo nel nostro ospedale era l’angiosarcoma post-radiazione. Questa predominanza di casi di cancro al seno spiega anche perché 11 dei 16 pazienti erano donne. C’era solo 1 caso di angiosarcoma associato a linfedema (Fig. 3), che coincide con i tassi di prevalenza riportati altrove. Il paziente aveva un linfedema cronico del braccio sinistro secondario alla dissezione linfonodale ascellare eseguita come parte del trattamento del cancro al seno 22 anni prima.
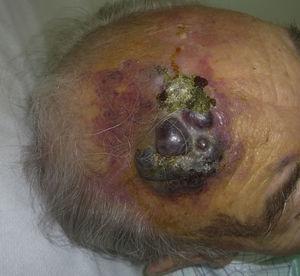
Placca rosso-violacea con aree nodulari sulla fronte di un maschio anziano.
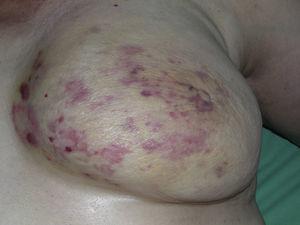
Molteplici macule e macchie eritematose con diverse papule rossastre su pelle giallastra su un seno irradiato a causa del cancro.
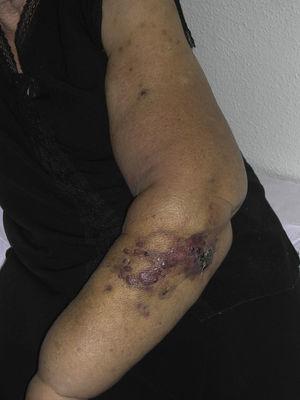
Papule e noduli rossastri-violacei con aree simili a lividi su un braccio con linfedema secondario a chirurgia del cancro al seno.
Con l’eccezione di 1 donna che aveva un angiosarcoma idiopatico o di Wilson-Jones, il resto delle donne della nostra serie (n=10) aveva una storia di cancro al seno. Nove di questi tumori erano carcinomi duttali invasivi. Poiché l’80% dei tumori al seno nella popolazione generale sono carcinomi duttali invasivi e solo il 10% sono carcinomi lobulari, l’alta prevalenza del carcinoma duttale invasivo nel nostro studio è probabilmente un riflesso dell’alta prevalenza di questo cancro nella popolazione generale, piuttosto che di una particolare associazione tra l’angiosarcoma cutaneo post-radiazione e il carcinoma duttale invasivo, come i nostri risultati sembrerebbero suggerire. In altre parole, le proporzioni osservate nella nostra serie sono in consonanza con i tassi descritti per diversi tumori del seno nella popolazione generale. C’erano 2 casi di angiosarcoma mammario inizialmente inclusi nel nostro studio che sono degni di menzione. È stato riportato che l’angiosarcoma mammario tende ad essere cutaneo quando è indotto da radiazioni e parenchimale quando non lo è.19 Nella nostra serie, dei 18 casi di angiosarcoma cutaneo inizialmente identificati, c’erano 2 casi di angiosarcoma mammario non indotto da radioterapia. Rivedendo questi casi, abbiamo scoperto che la sede primaria del tumore era il parenchima mammario e non la pelle. I tumori sono stati quindi esclusi dallo studio in quanto secondari e non primari. I casi rimanenti erano tutti angiosarcomi primari del seno indotti da radiazioni, in linea con i rapporti in letteratura.
La sede più comune per l’angiosarcoma nella nostra serie era il seno (10 casi) e non il viso o il cuoio capelluto, come ci si aspetterebbe. L’unico caso che ha coinvolto le estremità era un angiosarcoma associato a linfedema. Ancora una volta, il fatto che il sito tumorale più comune fosse il seno si spiegherebbe con la predominanza di tumori indotti da radiazioni nella nostra serie.
Anche se il periodo di latenza tra l’esposizione alla radioterapia e lo sviluppo dell’angiosarcoma è molto variabile secondo i rapporti in letteratura (3-50 anni), tende ad essere più lungo (in media, 25 anni) quando la malattia trattata da radiazioni è benigna. I periodi di latenza riportati per le malattie maligne sono più brevi (circa 10-15 anni), tranne nel caso dell’angiosarcoma del seno, per il quale è stata descritta una media di circa 5 anni.12 La ragione di questo periodo più breve nel caso dell’angiosarcoma della mammella non è chiara, sebbene siano state proposte diverse teorie, tra cui il grande volume di pelle irradiata, la presenza di un linfedema associato, fattori possibilmente intrinseci alla mammella e un possibile effetto sinergico con la chemioterapia.13,14 Nei nostri 10 casi post-radiazione, l’angiosarcoma è stato diagnosticato dopo una media di 8,2 anni e nel 90% dei casi sono passati almeno 5 anni. Non è stata riportata alcuna associazione tra periodo di latenza e prognosi per l’angiosarcoma. Nella nostra serie, il tempo medio dalla radioterapia allo sviluppo dell’angiosarcoma era un po’ più lungo nei pazienti morti (110,6 mesi) che in quelli sopravvissuti (82,25 mesi). Nell’unico caso di sindrome di Stewart-Treves, il paziente era stato sottoposto a dissezione linfonodale 22 anni prima. I periodi di latenza riportati per l’angiosarcoma associato al linfedema sono molto variabili, con una gamma da 1 a 30 anni e una media di 10. La sindrome di Stewart-Treves rappresenta il 90% di tutti i casi di angiosarcoma associato a linfedema.9,11 L’angiosarcoma può tuttavia insorgere anche in altre forme di linfedema, come il linfedema congenito, il linfedema filariale e il linfedema secondario alla dissezione linfonodale in altre parti del corpo.10
Le dimensioni del tumore sono attualmente il marker prognostico più ampiamente accettato per l’angiosarcoma cutaneo, ed è stato spesso riportato che gli angiosarcomi che misurano 5 cm o più hanno una prognosi peggiore rispetto ai tumori più piccoli.5,17 Nella nostra serie, abbiamo osservato differenze nelle dimensioni medie del tumore tra i sopravvissuti e i non sopravvissuti. Quelli che sono sopravvissuti avevano una dimensione media del tumore di 3,6 cm, che era oltre 3 volte più piccola della dimensione media (13,1 cm) nel gruppo di pazienti che sono morti. I nostri risultati supportano quindi la convinzione che i tumori più grandi sono associati a esiti peggiori nell’angiosarcoma e sottolineano l’importanza di diagnosticare il tumore quando inizia come una lesione simile a un livido, anche se questo è particolarmente difficile negli angiosarcomi del cuoio capelluto e ancor più quando il paziente ha ancora i capelli (Fig. 4). L’angiosarcoma della mammella post-radiazione è più facile da diagnosticare, poiché qualsiasi lesione persistente in quest’area con un aspetto vascolare o simile a un livido dovrebbe essere sottoposta a biopsia. Un indizio diagnostico potenzialmente utile è un alone giallastro (corrispondente all’emosiderina) intorno alla lesione sospetta (Fig. 5), poiché non abbiamo mai osservato questo segno nelle proliferazioni vascolari benigne sulla pelle irradiata. L’angiosarcoma post-radiazione e le proliferazioni vascolari atipiche su pelle irradiata devono essere distinte istologicamente, ma le caratteristiche cliniche possono aiutare. Le proliferazioni vascolari atipiche tendono ad essere molto più piccole degli angiosarcomi e generalmente hanno anche un periodo di latenza più breve.20 A differenza degli angiosarcomi, queste proliferazioni sono confinate al derma superficiale e medio e non invadono il tessuto sottocutaneo. Inoltre, l’istologia non mostra la caratteristica atipia nucleare osservata nell’angiosarcoma, né gli strati multipli di cellule endoteliali o le figure mitotiche. Tuttavia, occasionalmente può essere difficile distinguere tra le 2 entità e ci sono stati rapporti di coesistenza delle 2 lesioni nella stessa mammella irradiata. Ci sono stati anche casi di proliferazioni vascolari atipiche indotte da radiazioni che progrediscono in angiosarcoma.21 In casi molto complicati, può essere utile cercare la sovraespressione del gene MYC se viene eseguito uno studio immunoistochimico, o l’amplificazione se viene usata l’ibridazione fluorescente in situ. L’amplificazione di MYC è abbastanza comune negli angiosarcomi secondari, ma non sembra verificarsi nelle proliferazioni vascolari atipiche sulla pelle irradiata.22
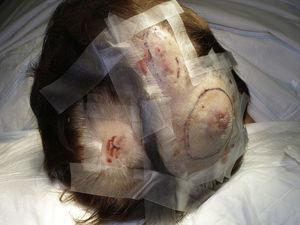
Noduli eritematosi multifocali sul cuoio capelluto.
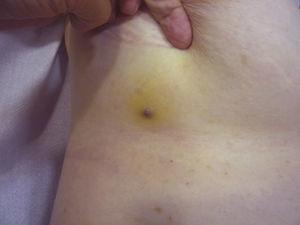
Papula rossastra-violacea che misura meno di 1cm circondata da un’area giallastra della pelle.
L’altro marker prognostico più ampiamente accettato nell’angiosarcoma è l’età. Nella nostra serie, i pazienti che sono sopravvissuti erano in media più giovani di quelli che sono morti (62 contro 71 anni). Ciononostante, 10 dei 16 pazienti della nostra serie erano morti entro la fine dello studio, confermando la cattiva prognosi generalmente associata all’angiosarcoma.
A differenza del caso di altri sarcomi, l’American Joint Committee on Cancer non considera il grado istologico un marker prognostico rilevante nell’angiosarcoma. Diversi autori, tuttavia, hanno suggerito che alcune caratteristiche istopatologiche potrebbero avere un peso sulla prognosi. Alcuni studi recenti, per esempio, hanno sostenuto che un modello solido predominante potrebbe essere un marcatore prognostico relativamente buono nell’angiosarcoma della testa e del collo.3,16 Questo non era il caso nella nostra serie, dato che 6 dei 10 pazienti che sono morti avevano questo pattern rispetto a solo 2 dei 6 pazienti che erano ancora vivi alla fine dello studio (Figg. 6 e 7).
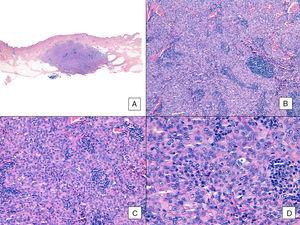
Angiosarcoma con un modello solido predominante. A, l’immagine panoramica mostra l’invasione del derma reticolare medio e profondo e dell’ipoderma (ematossilina-eosina, ingrandimento originale ×10). B, tumore densamente cellulare che ha distrutto le strutture preesistenti ed è accompagnato da infiltrati linfoidi nodulari (ematossilina-eosina, ingrandimento originale ×100). C e D, vista dettagliata che mostra una predominanza di cellule epitelioidi accompagnate da un infiltrato linfocitario (ematossilina-eosina ×200 e ×400, rispettivamente).
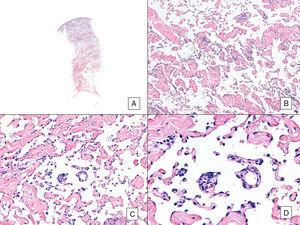
Angiosarcoma con un modello vasoformativo predominante. A, immagine panoramica che mostra l’infiltrazione del derma fino all’ipoderma (ematossilina-eosina, ingrandimento originale ×10). B, spazi vascolari neoplastici con pronunciata dissezione del collagene (ematossilina-eosina, ingrandimento originale ×100). C e D, vasi non neoplastici sezionati da cellule endoteliali neoplastiche, che rimangono “galleggianti” nel derma (segno promontorio) (ematossilina-eosina, ingrandimento originale ×200 e ×400, rispettivamente).
Altri fattori istologici potenzialmente ad alto rischio proposti in letteratura sono la presenza di necrosi, una morfologia epitelioide e una maggiore profondità di invasione.5,7 I nostri risultati supportano questi potenziali marcatori, poiché tutte e 3 le variabili erano più comuni nel gruppo dei non sopravvissuti. La necrosi è stata osservata in 5 dei 10 pazienti morti e in solo 1 dei 6 pazienti sopravvissuti. Allo stesso modo, gli unici 2 pazienti con un modello a cellule fusate predominante erano ancora vivi alla fine dello studio. Nessuno dei pazienti che sono morti, al contrario, aveva questo pattern. Infine, gli unici 2 pazienti con coinvolgimento dei piani muscolari sono morti. Il numero medio di mitosi era anche più alto nel gruppo dei non sopravvissuti (18.3 vs 10).
Il trattamento di scelta dell’angiosarcoma cutaneo è l’escissione chirurgica con ampi margini seguita da radioterapia.6 La frequente natura multifocale dell’angiosarcoma e la sua propensione a sviluppare diffusione subclinica spesso complica il raggiungimento di margini chiari. Inoltre, non esiste un accordo universale su ciò che costituisce un margine adeguato, e la maggior parte degli studi fornisce informazioni imprecise, come l’escissione con “ampi margini”. Nella nostra serie, la chirurgia è stata il trattamento di scelta in 14 dei 16 pazienti. Sono stati utilizzati margini di 3 cm quando era tecnicamente possibile, e negli altri casi si è tentato di eliminare il tumore con margini di 2 cm. I 14 pazienti sono stati sottoposti a chirurgia radicale. Gli altri 2 non sono stati ritenuti candidati alla chirurgia e hanno ricevuto una chemioterapia palliativa. Sono morti durante il follow-up. La radioterapia adiuvante al letto chirurgico è stata utilizzata solo in 4 pazienti. Questo basso uso della radioterapia nella gestione dell’angiosarcoma nella nostra serie può probabilmente essere spiegato dal gran numero di casi di angiosarcomi post-radiazione nella nostra serie. In altre parole, è stato probabilmente influenzato da un certo livello di resistenza all’uso delle radiazioni in questi casi. L’uso della radioterapia nell’angiosarcoma post-radiazione, tuttavia, trova sostegno nella letteratura, e talvolta viene persino somministrata come monoterapia, senza chirurgia.23,24 La chemioterapia è stata utilizzata nella metà dei pazienti. Come già accennato, aveva un ruolo meramente palliativo ed era associata ad un cattivo esito in tutti i casi. Anche se la monoterapia con paclitaxel non era il regime di chemioterapia più utilizzato nella nostra serie (poiché i casi sono stati diagnosticati qualche tempo fa), questo regime è ora utilizzato come opzione di prima linea nella maggior parte dei casi. Nonostante le aspettative iniziali, i farmaci angiogenici (sunitinib, sorafenib, bevacizumab, talidomide) non sono utilizzati in questo contesto a causa dei loro risultati deludenti.
Nonostante il piccolo numero di casi nella nostra serie di angiosarcoma cutaneo, che ci ha impedito di eseguire analisi statistiche, abbiamo osservato che le dimensioni più grandi del tumore e l’età più avanzata erano entrambi associati a una prognosi peggiore. Un’associazione meno evidente è stata osservata anche tra la prognosi sfavorevole e le seguenti caratteristiche istologiche: presenza di necrosi, una predominanza di cellule epitelioidi, invasione degli strati più profondi e un maggior numero di mitosi.
Discrezioni eticheProtezione dell’uomo e degli animali
Gli autori dichiarano che nessun test è stato effettuato su esseri umani o animali ai fini di questo studio.
Confidenzialità dei dati
Gli autori dichiarano di aver seguito il protocollo del loro ospedale sulla pubblicazione dei dati riguardanti i pazienti.
Diritto alla privacy e al consenso informato
Gli autori dichiarano che nessun dato privato dei pazienti appare in questo articolo.
Conflitti di interesse
Gli autori dichiarano di non avere conflitti di interesse.
Gli autori dichiarano di non avere conflitti di interesse.