L’angiosarcome cutané a l’un des pires pronostics parmi toutes les tumeurs cutanées. Il est très agressif et présente des taux de récidive locale élevés. Le taux de survie à 5 ans se situe entre 12% et 34% selon la plupart des études,1,2 mais il peut atteindre 62%.3 Contrairement à d’autres sarcomes, le degré de différenciation n’a pas été associé au pronostic de l’angiosarcome cutané.4
La forme classique de l’angiosarcome cutané est une lésion œdémateuse mal définie, ressemblant à une ecchymose, avec une présentation clinique largement indolente dans ses premières phases. Elle se produit sur le visage ou le cuir chevelu des patients âgés (angiosarcome de Wilson-Jones) et représente environ 50 % de tous les angiosarcomes cutanés primaires.5-8 Deux autres formes typiques d’angiosarcome sont le syndrome de Stewart-Treves, qui se développe dans les zones de lymphoedème de longue date et est particulièrement fréquent chez les femmes ayant subi une mastectomie radicale,9-11 et l’angiosarcome post-radiation, qui se développe dans les zones de peau irradiée, en particulier dans la région pectorale des femmes ayant des antécédents de cancer du sein traité par radiothérapie12.-15
L’aspect histopathologique de l’angiosarcome cutané varie de formes relativement différenciées avec des espaces vasculaires reconnaissables couverts par des cellules endothéliales proéminentes avec quelques atypies et un motif infiltrant disséquant les faisceaux de collagène à des formes plus solides hautement indifférenciées composées de cellules fusiformes ou épithélioïdes avec des atypies et un pléomorphisme considérablement plus importants et un nombre plus élevé de mitoses. Les espaces vasculaires sont rares et les tumeurs peuvent parfois ressembler à des carcinomes.
Le traitement de base des angiosarcomes cutanés – et le seul qui soit potentiellement curatif si des marges sans maladie sont obtenues – est l’excision chirurgicale avec de larges marges suivie d’une radiothérapie locale et même, selon certains auteurs, d’une irradiation des ganglions lymphatiques régionaux.6 Dans la plupart des cas, cependant, il n’est pas facile d’obtenir des marges sans maladie en raison d’une propagation subclinique étendue. En outre, ces tumeurs sont fréquemment multifocales. La chimiothérapie a un rôle purement palliatif dans la prise en charge de l’angiosarcome cutané.
Bien que l’angiosarcome cutané soit rare (il représente moins de 1% de tous les sarcomes), la plupart des cas d’angiosarcomes proviennent de la peau. En raison de leur faible prévalence, les angiosarcomes cutanés sont inclus dans des séries d’angiosarcomes viscéraux ou osseux, qui ont un pronostic encore plus sombre.1 Il y a donc peu de grandes séries d’angiosarcomes cutanés dans la littérature en raison du manque de cas uniformes à long terme.2,4,5,7,16 La gestion des angiosarcomes est en outre souvent décourageante, surtout dans les cas de maladie avancée, qui ont un très mauvais pronostic malgré l’utilisation d’un traitement agressif dès le début. Motivés par les difficultés associées à la gestion de l’angiosarcome cutané et par le peu de littérature disponible, nous avons étudié tous les cas d’angiosarcome cutané traités à l’Instituto Valenciano de Oncología (IVO), à Valence, en Espagne, dans le but d’identifier les facteurs cliniques, histologiques et liés au traitement qui pourraient être associés au pronostic. Pour ce faire, nous avons examiné les dossiers médicaux et les résultats cliniques à la recherche de données exploratoires qui pourraient servir de guide pour un diagnostic précoce, car les patients présentant une maladie précoce et de petites tumeurs ont une probabilité de survie considérablement améliorée.
Matériel et méthodes
Nous avons mené une étude observationnelle rétrospective de tous les cas d’angiosarcomes cutanés traités à l’IVO entre janvier 2000 et décembre 2015. Toutes les informations compilées ont été extraites des dossiers médicaux des patients, des archives des biopsies du service de pathologie et des archives photographiques de notre service. Sur les 20 cas initialement identifiés, 4 ont dû être exclus : 1 parce qu’il n’y avait pas de suivi, un autre parce que le matériel était insuffisant pour déterminer si la tumeur était un hémangioendothéliome ou un angiosarcome, et 2 parce que les tumeurs n’étaient pas des angiosarcomes primaires. Ces deux tumeurs avaient initialement été étiquetées comme angiosarcomes cutanés parce que toutes les lames d’histologie montraient une atteinte cutanée du sein. Cependant, en révisant les blocs, nous avons constaté que l’atteinte cutanée était secondaire dans les 2 cas, et que la tumeur primaire était située dans le parenchyme mammaire d’où elle s’étendait jusqu’à la peau sus-jacente.
Les critères d’inclusion de l’étude étaient des résultats cliniques évocateurs d’un angiosarcome cutané et la confirmation histologique du diagnostic par une coloration à l’hématoxyline-éosine des spécimens de biopsie, appuyée par des études immunohistochimiques, qui dans la plupart des cas impliquaient une coloration CD31, CD34, D240 et Ki-67.
Les variables suivantes ont été étudiées pour chaque patient : âge, sexe, localisation et taille de la tumeur, type d’angiosarcome (primaire, post-radiation, associé à un lymphœdème), traitement (chirurgie, radiothérapie, chimiothérapie), récidive, métastases, survie et décès. Pour les tumeurs post-radiation et associées à un lymphœdème, nous avons également enregistré le type de tumeur antérieure et le nombre d’années écoulées depuis la radiothérapie ou le lymphœdème. Les variables histologiques analysées étaient le statut de la marge, le schéma histopathologique (vasoformatif, solide ou mixte), le type cellulaire prédominant (épithélioïde ou à cellules fusiformes), la présence de nécrose (oui, non), le niveau d’invasion (épiderme, derme, hypoderme, muscle, os), la réaction lymphocytaire, le schéma d’infiltration et le nombre de mitoses par 10 champs.
Résultats
Sixteen cas d’angiosarcomes cutanés ont été inclus dans l’étude. Ils correspondaient à 11 femmes et 5 hommes âgés de 35 à 83 ans (moyenne, 67 ans ; médiane, 71 ans). Dix des cas étaient des angiosarcomes post-radiation (10 cas), 5 des angiosarcomes de Wilson-Jones et un seul des angiosarcomes associés à un lymphœdème. La localisation la plus fréquente était le tronc (10 cas), suivi de la tête et du cou (5 cas). Les membres supérieurs n’ont été touchés que dans un seul cas. La plus petite taille de la tumeur était de 1cm et la plus grande de 50cm (moyenne, 10cm ; médiane, 6,5cm).
Onze des patients avaient des antécédents de cancer (cancer du sein dans 10 cas et séminome dans 1 cas). A l’exception d’un cas de carcinome lobulaire invasif, tous les cancers du sein étaient des carcinomes canalaires invasifs.
Le délai moyen entre la radiothérapie et le développement de l’angiosarcome dans les 10 cas d’angiosarcome post-radiation était de 8,2 ans. Un seul des cas est apparu dans les 5 ans suivant la radiothérapie ; les autres sont apparus au moins 5 ans plus tard.
Quatorze cas ont été traités chirurgicalement et une radiothérapie adjuvante a été utilisée dans 4 d’entre eux. Huit patients ont reçu une chimiothérapie et c’était le premier et seul traitement chez 2 patients.
La doxorubicine et le taxol ont été utilisés chacun dans 4 cas, l’ifosfamide a été utilisé dans 3 cas et le paclitaxel et la dacarbazine ont été utilisés dans 1 cas chacun. La réponse à la chimiothérapie était faible, et bien que presque tous les patients aient présenté une réponse partielle, la maladie a progressé dans tous les cas et les patients sont décédés au cours du suivi (8/8).
Cinq patients présentaient des métastases à distance, qui impliquaient plusieurs sites dans la plupart des cas. Les sites les plus fréquents étaient le poumon et le foie.
Dix des 16 patients sont décédés d’angiosarcome au cours du suivi. Les 6 autres patients sont actuellement exempts de la maladie. La durée moyenne du suivi était de 42,5 mois (médiane, 26 mois ; intervalle, 7-188 mois).
Histologiquement, 8 cas présentaient un schéma de croissance solide, 4 un schéma vasoformatif et 4 un schéma mixte. Le type cellulaire prédominant était épithélioïde dans 14 cas et fusiforme dans seulement 2. Une nécrose a été observée dans 6 tumeurs et le schéma d’infiltration était sous-cutané dans la plupart des cas (n=10). Quatre cas étaient confinés au derme et seulement 2 touchaient les plans musculaires. Les marges chirurgicales n’étaient pas évaluables dans 3 cas. Parmi les autres cas, 8 avaient des marges négatives et 5 des marges positives. La réaction lymphocytaire était légère ou modérée dans 10 cas, intense dans 2 cas, et inexistante dans 4 cas. Dans les 14 cas avec une réaction lymphocytaire, l’infiltrat était péritumoral dans 2 cas, intratumoral dans 8 cas, et mixte dans 2 cas. Il y avait une moyenne de 15 mitoses par 10 champs (gamme, 0-37 mitoses).
Les résultats cliniques et pathologiques les plus pertinents sont résumés dans le tableau 1. Les résultats de la comparaison entre les survivants et les non-survivants sont résumés dans le tableau 2.
Sélection de résultats cliniques et pathologiques pour les 16 angiosarcomes cutanés.a
| Patient | Age, y | Sexe | Type | Localisation | Taille, cm | Durée depuis Rx, mo | Dose, Gy | Tumeur antérieure | Type de cancer du sein | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | F | PR | Sein gauche | 18 | 60 | 46 | Sein | IDC | |
| 2 | 71 | F | ST | Bras gauche | 12 | – | – | Sein | IDC | |
| 3 | 51 | F | PR | Région sous-mammaire droite | 1 | 57 | 50 | Sein | IDC | |
| 4 | 76 | F | WJ | Tête et cou | 3 | – | – | Non | . | |
| 5 | 77 | F | PR | sein droit | 1 | 94 | 46 | Sein | IDC | |
| 6 | 71 | F | PR | Sein gauche | 50 | 171 | 48 | Sein | IDC | |
| 7 | 48 | M | PR | Paroi abdominale | 2 | 96 | 26 | Seminome | – | |
| 8 | 55 | F | PR | Poitrine gauche | 10 | 88 | 46 | Sein | ILC | |
| 9 | 69 | F | PR | Poitrine gauche | 8 | 143 | 46 | Poitrine | IDC | |
| 10 | . 76 | M | WJ | Joue droite | 6 | – | – | No | – | |
| 11 | 35 | F | PR | Mammaire droit | 12 | 66 | 50 | Poitrine | IDC | |
| 12 | 57 | F | PR | . Poitrine droite | 8 | 108 | 50 | Poitrine | IDC | |
| 13 | . | 68 | M | WJ | Tête et cou | 2 | – | – | Non | – |
| 14 | 80 | M | WJ | Tête et cou | 15 | – | – | Non | – | |
| 15 | 79 | M | WJ | Tête et cou | 2 | – | – | Non | – | |
| 16 | 83 | F | PR | Poitrine gauche | 3 | 110 | 50 | Poitrine | IDC | |
| X=67.1 | 11W, 5M | 10 RI, 5 WJ, 1 ST | 9 poitrine, 5 tête et cou, 1 abdomen, 1 membre supérieur | X=10 | X=100.3 | X=45.8 | 10 seins, 1 seminome |
10 cancers du sein : 9 IDC, 1 ILC |
| Patient | Chirurgie | Marge, cm | Traitement AS | Décès | Modèle HP | Type de cellule | Nécrose | DoI | Mitoses/mm2 | Survie, mo |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Oui | 0.2 | 13 | Oui | 1 | E | Non | 3 | 7 | 24 |
| 2 | Non | ns | 3 | Oui | 2 | E | Non | 3 | 37 | 8 |
| 3 | Oui | 3.5 | 1 | Non | 1 | E | Non | 3 | 2 | 29 |
| 4 | Oui | 2 | 123 | Oui | 2 | E | Non | 3 | 28 | 26 |
| 5 | Oui | ns | 1 | Oui | 2 | E | Non | 2 | 14 | 8 |
| 6 | Non | ns | 3 | Oui | 1 | E | Non | 2 | 0 | 19 |
| 7 | Oui | ns | 1 | Non | 3 | E | Non | 3 | 6 | 187 |
| 8 | Oui | ns | 13 | Oui | 2 | E | Oui | 4 | 6 | 28 |
| 9 | Oui | ns | 13 | Oui | 3 | E | Oui | 3 | 22 | 24 |
| 10 | Oui | ns | 12 | Oui | 2 | E | Oui | 3 | 16 | 7 |
| 11 | Oui | 0.5 | 1 | Non | 2 | SC | Non | 3 | 18 | 76 |
| 12 | Oui | 0 | 13 | Oui | 3 | E | Oui | 3 | 36 | 26 |
| 13 | Oui | 2 | 12 | Non | 3 | E | Non | 3 | 5 | 95 |
| 14 | Oui | 2 | 123 | Oui | 2 | E | Oui | 4 | 17 | 24 |
| 15 | Oui | 2 | 1 | Non | 2 | E | Oui | 2 | 6 | 53 |
| 16 | Oui | 3 | 1 | Non | 1 | SC | Non | 2 | 23 | 51 |
| 14 oui, 2 non | 1.68 | 14 : Qx 4 : Rt 8 : Qt |
10 oui 6 non |
4 vasof 8 solide 4 mixte |
14 E, 2 SC | 6 oui 10 non |
4 derme 10 hypoderme 2 muscle |
X=15 | X=42.8 |
Les patients décédés sont indiqués en gras.
Traitement AS, traitement de l’angiosarcome (1, chirurgie ; 2, radiothérapie ; 3, chimiothérapie ) ; DoI, profondeur d’invasion (2, derme ; 3, hypoderme ; 4, muscle) ; E, épithélioïde ; HP, histopathologique (1, vasoformatif ; 2,solide ; 3, mixte) ; IDC, carcinome canalaire invasif ; ILC, carcinome lobulaire invasif ; M, homme ; ns, non spécifié ; PR, angiosarcome post-radiation ; Rx, radiation ; SC, cellule fusiforme ; ST, angiosarcome de Stewart-Treves ; W, femme ; WJ, angiosarcome de Wilson-Jones : X, moyenne.
Comparaison des variables entre les survivants et les patients décédés d’un angiosarcome cutané.
| Variable | Vivants (n=6) | Décédés (n=10) |
|---|---|---|
| Age, moyenne, y | 61 | 71 |
| Femmes | 3 | 8 |
| Hommes | 3 | 2 |
| Postradiation | 4 | 6 |
| Idiopathique | 2 | 3 |
| Lymphœdème-associé | 0 | 1 |
| Tronc | 4 | 6 |
| Tête et cou | 2 | 3 |
| Membres supérieurs | 0 | 1 |
| Taille, cm | 3.6 | 13,1 |
| Temps depuis la radiothérapie, mo | 82,25 | 110.6 |
| Dose de radiation, Gy | 44 | 47 |
| Sein | 3 | 7 |
| Seminome | 1 | |
| Chirurgie | 6 | 8 |
| Radiothérapie | 1 | 3 |
| Chimiothérapie | 0 | 8 |
| Vasoformative | 2 | 2 |
| Solide | 2 | 6 |
| Mixte | 2 | 2 |
| Nécrose | 1 | 5 |
| Dermique | 2 | 2 |
| Sous-cutané | 4 | 6 |
| Muscle | 0 | 2 |
| Mitoses | 10 | 18.3 |
| Survie, mo | 81,8 | 19.4 |
Discussion
L’angiosarcome cutané est une tumeur très rare, comme en témoigne le fait que nous n’avons pu recueillir des données que sur 16 tumeurs diagnostiquées sur une période de 14 ans dans un hôpital d’oncologie. Globalement, l’angiosarcome cutané est un peu plus fréquent chez les hommes âgés. Cela s’explique par le fait que la forme la plus courante d’angiosarcome cutané dans la population générale est l’angiosarcome primaire de la tête et du cou (également connu sous le nom d’angiosarcome idiopathique ou de Wilson-Jones) (Fig. 1), qui a tendance à toucher les hommes âgés5,7,17. L’angiosarcome post-radiation (Fig. 2) est désormais la deuxième forme la plus fréquente d’angiosarcome cutané en raison de l’utilisation accrue de la radiothérapie par rapport à la mastectomie radicale pour traiter le cancer du sein.2,13,18 Ce changement a également conduit à une réduction de la fréquence de l’angiosarcome associé au lymphœdème, qui est actuellement la forme la moins fréquente de cette tumeur. La prévalence des différentes formes d’angiosarcome cutané dans notre série ne coïncide pas avec les rapports de la littérature car notre hôpital traite un grand nombre de femmes atteintes de cancer du sein, ce qui explique que la forme la plus fréquente d’angiosarcome cutané dans notre hôpital était l’angiosarcome post-radiation. Cette prédominance des cas de cancer du sein explique également pourquoi 11 des 16 patients étaient des femmes. Il n’y avait qu’un seul cas d’angiosarcome associé à un lymphœdème (figure 3), ce qui coïncide avec les taux de prévalence rapportés ailleurs. La patiente présentait un lymphœdème chronique du bras gauche secondaire à un curage ganglionnaire axillaire effectué dans le cadre du traitement du cancer du sein 22 ans auparavant.
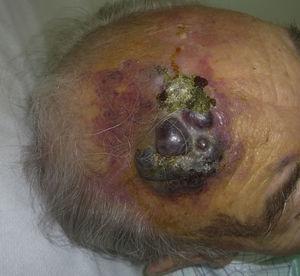
Plaque rouge-violacée avec des zones nodulaires sur le front d’un homme âgé.
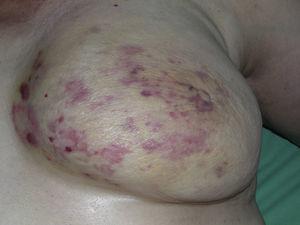
Multiples macules et plaques érythémateuses avec plusieurs papules rougeâtres sur une peau jaunâtre sur un sein irradié en raison d’un cancer.
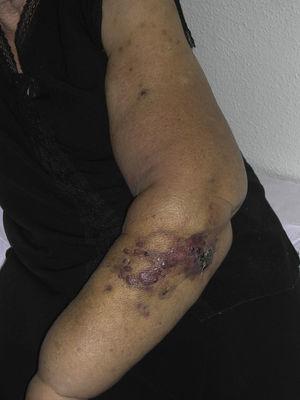
Papules et nodules rougeâtres-violacés avec des zones ressemblant à des ecchymoses sur un bras présentant un lymphoedème secondaire à une chirurgie du cancer du sein.
À l’exception d’une femme qui avait un angiosarcome idiopathique ou de Wilson-Jones, le reste des femmes de notre série (n=10) avait des antécédents de cancer du sein. Neuf de ces cancers étaient des carcinomes canalaires invasifs. Comme 80% des cancers du sein dans la population générale sont des carcinomes canalaires invasifs et seulement 10% des carcinomes lobulaires, la forte prévalence du carcinome canalaire invasif dans notre étude est probablement le reflet de la forte prévalence de ce cancer dans la population générale, plutôt que d’une association particulière entre angiosarcome cutané post-radique et carcinome canalaire invasif, comme nos résultats semblent le suggérer. En d’autres termes, les proportions observées dans notre série sont en consonance avec les taux décrits pour les différents cancers du sein dans la population générale. Deux cas d’angiosarcome du sein initialement inclus dans notre étude méritent d’être mentionnés. Il a été rapporté que l’angiosarcome du sein tend à être cutané lorsqu’il est induit par la radiothérapie et parenchymateux lorsqu’il ne l’est pas.19 Dans notre série, sur les 18 cas d’angiosarcome cutané initialement identifiés, il y avait 2 cas d’angiosarcome du sein non induits par la radiothérapie. En examinant ces cas, nous avons constaté que la localisation primaire de la tumeur était le parenchyme mammaire et non la peau. Ces tumeurs ont donc été exclues de l’étude car elles étaient secondaires et non primaires. Les autres cas étaient tous des angiosarcomes primaires du sein induits par des radiations, conformément aux rapports de la littérature.
La localisation la plus fréquente de l’angiosarcome dans notre série était le sein (10 cas) et non le visage ou le cuir chevelu, comme on pouvait s’y attendre. Le seul cas qui a touché les extrémités était un angiosarcome associé à un lymphœdème. Là encore, le fait que le site tumoral le plus fréquent était le sein s’expliquerait par la prédominance des tumeurs radio-induites dans notre série.
Bien que la période de latence entre l’exposition à la radiothérapie et le développement d’un angiosarcome soit très variable selon les rapports de la littérature (3-50 ans), elle tend à être plus longue (en moyenne 25 ans) lorsque la maladie traitée par radiation est bénigne. Les périodes de latence rapportées pour les maladies malignes sont plus courtes (environ 10-15 ans), sauf dans le cas de l’angiosarcome du sein, pour lequel une moyenne d’environ 5 ans a été décrite12. La raison de cette période plus courte dans le cas de l’angiosarcome du sein n’est pas claire, bien que plusieurs théories aient été proposées, y compris le grand volume de peau irradiée, la présence d’un lymphœdème associé, des facteurs qui sont peut-être intrinsèques au sein, et un possible effet synergique avec la chimiothérapie.13,14 Dans nos 10 cas post-radiation, l’angiosarcome a été diagnostiqué après une moyenne de 8,2 ans, et au moins 5 ans s’étaient écoulés dans 90% des cas. Aucune association entre la période de latence et le pronostic n’a été rapportée pour l’angiosarcome. Dans notre série, le délai moyen entre la radiothérapie et le développement de l’angiosarcome était un peu plus long chez les patients décédés (110,6 mois) que chez ceux qui ont survécu (82,25 mois). Dans le seul cas du syndrome de Stewart-Treves, le patient avait subi un curage ganglionnaire 22 ans auparavant. Les périodes de latence rapportées pour les angiosarcomes associés à un lymphœdème sont très variables, avec une fourchette de 1 à 30 ans et une moyenne de 10 ans. Le syndrome de Stewart-Treves représente 90 % de tous les cas d’angiosarcome associé à un lymphœdème9,11. Cependant, l’angiosarcome peut également survenir dans d’autres formes de lymphœdème, comme le lymphœdème congénital, le lymphœdème filarien et le lymphœdème secondaire à une dissection des ganglions lymphatiques dans d’autres parties du corps10.
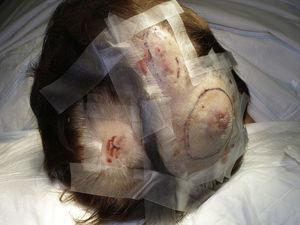
La taille de la tumeur est actuellement le marqueur pronostique le plus largement accepté pour l’angiosarcome cutané, et il a été fréquemment rapporté que les angiosarcomes mesurant 5cm ou plus ont un pronostic plus mauvais que les tumeurs plus petites.5,17 Dans notre série, nous avons observé des différences dans la taille moyenne de la tumeur entre les survivants et les non-survivants. Ceux qui ont survécu avaient une taille moyenne de tumeur de 3,6 cm, ce qui était plus de 3 fois plus petit que la taille moyenne (13,1 cm) dans le groupe de patients qui sont décédés. Nos résultats confirment donc la croyance selon laquelle les tumeurs de grande taille sont associées à de plus mauvais résultats dans les angiosarcomes, et soulignent l’importance de diagnostiquer la tumeur lorsqu’elle commence par une lésion ressemblant à une ecchymose, bien que cela soit particulièrement difficile dans les angiosarcomes du cuir chevelu et encore plus lorsque le patient a encore des cheveux (Fig. 4). L’angiosarcome du sein post-radiation est plus facile à diagnostiquer, car toute lésion persistante dans cette région ayant un aspect vasculaire ou d’ecchymose doit être biopsiée. Un indice diagnostique potentiellement utile est un halo jaunâtre (correspondant à l’hémosidérine) autour de la lésion suspecte (Fig. 5), car nous n’avons jamais observé ce signe dans les proliférations vasculaires bénignes sur la peau irradiée. L’angiosarcome post-radique et les proliférations vasculaires atypiques sur peau irradiée doivent être distingués histologiquement mais les caractéristiques cliniques peuvent aider. Les proliférations vasculaires atypiques ont tendance à être beaucoup plus petites que les angiosarcomes et leur période de latence est généralement plus courte.20 Contrairement aux angiosarcomes, ces proliférations sont confinées au derme superficiel et moyen et n’envahissent pas le tissu sous-cutané. En outre, l’histologie ne présente pas les atypies nucléaires caractéristiques observées dans les angiosarcomes, ni les multiples couches de cellules endothéliales ou les figures mitotiques. Néanmoins, il peut parfois être difficile de distinguer les deux entités et des cas de coexistence des deux lésions dans un même sein irradié ont été rapportés. Il y a même eu des cas de proliférations vasculaires atypiques radio-induites évoluant vers un angiosarcome.21 Dans les cas très compliqués, il peut être utile de rechercher une surexpression du gène MYC si une étude immunohistochimique est réalisée, ou une amplification si une hybridation in situ fluorescente est utilisée. L’amplification de MYC est assez fréquente dans les angiosarcomes secondaires, mais ne semble pas se produire dans les proliférations vasculaires atypiques sur peau irradiée.22
Nodules érythémateux multifocaux sur le cuir chevelu.
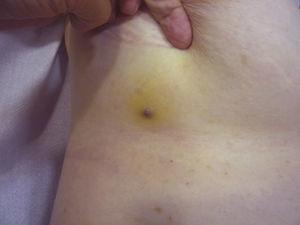
Papule rougeâtre-violacée mesurant moins de 1cm entourée d’une zone jaunâtre de la peau.
L’autre marqueur pronostique le plus largement accepté dans l’angiosarcome est l’âge. Dans notre série, les patients qui ont survécu étaient en moyenne plus jeunes que ceux qui sont décédés (62 contre 71 ans). Néanmoins, 10 des 16 patients de notre série étaient décédés à la fin de l’étude, ce qui corrobore le mauvais pronostic généralement associé à l’angiosarcome.
Contrairement au cas des autres sarcomes, l’American Joint Committee on Cancer ne considère pas le grade histologique comme un marqueur pronostique pertinent dans l’angiosarcome. Plusieurs auteurs ont cependant suggéré que certaines caractéristiques histopathologiques pourraient avoir un impact sur le pronostic. Certaines études récentes, par exemple, ont affirmé qu’un aspect solide prédominant pourrait être un marqueur de pronostic relativement bon dans les angiosarcomes de la tête et du cou3,16. Cela n’a pas été le cas dans notre série puisque 6 des 10 patients décédés présentaient ce motif contre seulement 2 des 6 patients encore en vie à la fin de l’étude (figures 6 et 7).
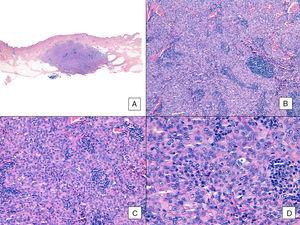
Angiosarcome avec un motif solide prédominant. A, L’image panoramique montre une invasion du derme réticulaire moyen et profond et de l’hypoderme (hématoxyline-éosine, grossissement original ×10). B, Tumeur densément cellulaire ayant détruit les structures préexistantes et accompagnée d’infiltrats lymphoïdes nodulaires (hématoxyline-éosine, grossissement original ×100). C et D, Vue détaillée montrant une prédominance de cellules épithélioïdes accompagnées d’un infiltrat lymphocytaire (hématoxyline-éosine ×200 et ×400, respectivement).
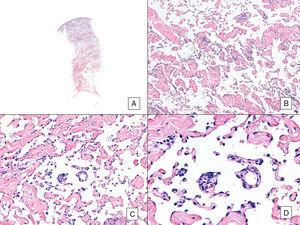
Angiosarcome avec un motif vasoformatif prédominant. A, image panoramique montrant une infiltration du derme jusqu’à l’hypoderme (hématoxyline-éosine, grossissement original ×10). B, espaces vasculaires néoplasiques avec dissection prononcée du collagène (hématoxyline-éosine, grossissement original ×100). C et D, vaisseaux non néoplasiques disséqués par des cellules endothéliales néoplasiques, qui restent « flottantes » dans le derme (signe du promontoire) (hématoxyline-éosine, grossissement original ×200 et ×400, respectivement).
D’autres facteurs histologiques potentiellement à haut risque proposés dans la littérature sont la présence de nécrose, une morphologie épithélioïde et une plus grande profondeur d’invasion5,7. Nos résultats confirment ces marqueurs potentiels, car les 3 variables étaient plus fréquentes dans le groupe des non-survivants. La nécrose a été observée chez 5 des 10 patients qui sont décédés et chez seulement 1 des 6 patients qui ont survécu. De même, les deux seuls patients présentant une prédominance de cellules fusiformes étaient encore en vie à la fin de l’étude. En revanche, aucun des patients décédés ne présentait ce profil. Enfin, les 2 seuls patients présentant une atteinte des plans musculaires sont décédés. Le nombre moyen de mitoses était également plus élevé dans le groupe des non survivants (18,3 vs 10).
Le traitement de choix de l’angiosarcome cutané est l’excision chirurgicale avec de larges marges suivie d’une radiothérapie.6 La nature multifocale fréquente de l’angiosarcome et sa propension à développer une propagation subclinique compliquent souvent l’obtention de marges nettes. En outre, il n’existe pas d’accord universel sur ce qui constitue une marge adéquate, et la plupart des études fournissent des informations imprécises, telles que l’excision avec des « marges larges ». Dans notre série, la chirurgie a été le traitement de choix chez 14 des 16 patients. Des marges de 3 cm ont été utilisées lorsque cela était techniquement possible, et dans les autres cas, l’élimination a été tentée en utilisant des marges de 2 cm. Les 14 patients ont subi une chirurgie radicale. Les 2 autres n’ont pas été jugés candidats à la chirurgie et ont reçu une chimiothérapie palliative. Ils sont décédés au cours du suivi. La radiothérapie adjuvante au lit chirurgical n’a été utilisée que chez 4 patients. Cette faible utilisation de la radiothérapie dans la gestion de l’angiosarcome dans notre série peut probablement s’expliquer par le grand nombre de cas d’angiosarcomes post-radiation dans notre série. En d’autres termes, elle a probablement été influencée par un certain niveau de résistance à l’utilisation de la radiothérapie dans ces cas. L’utilisation de la radiothérapie dans les angiosarcomes post-radiation est cependant soutenue dans la littérature, et est même parfois administrée en monothérapie, sans chirurgie.23,24 La chimiothérapie a été utilisée chez la moitié des patients. Comme nous l’avons déjà mentionné, elle avait un rôle purement palliatif et était associée à un mauvais résultat dans tous les cas. Bien que la monothérapie par paclitaxel n’ait pas été le régime de chimiothérapie le plus utilisé dans notre série (car les cas ont été diagnostiqués il y a un certain temps), ce régime est maintenant utilisé comme option de première ligne dans la plupart des cas. Malgré les attentes initiales générées, les médicaments angiogéniques (sunitinib, sorafenib, bevacizumab, thalidomide) ne sont pas utilisés dans ce contexte en raison de leurs résultats décevants.
Malgré le petit nombre de cas dans notre série d’angiosarcomes cutanés, qui nous a empêché de réaliser des analyses statistiques, nous avons observé que la taille plus importante de la tumeur et l’âge plus avancé étaient tous deux associés à un pronostic plus mauvais. Une association moins évidente a également été observée entre le mauvais pronostic et les caractéristiques histologiques suivantes : présence de nécrose, prédominance de cellules épithélioïdes, invasion des couches plus profondes et un plus grand nombre de mitoses.
Divulgations éthiquesProtection de l’homme et de l’animal
Les auteurs déclarent qu’aucun test n’a été effectué chez l’homme ou l’animal dans le cadre de cette étude.
Confidentialité des données
Les auteurs déclarent avoir suivi le protocole de leur hôpital sur la publication des données concernant les patients.
Droit à la vie privée et consentement éclairé
Les auteurs déclarent qu’aucune donnée privée de patient n’apparaît dans cet article.
Conflits d’intérêts
Les auteurs déclarent n’avoir aucun conflit d’intérêts.