El angiosarcoma cutáneo tiene uno de los peores pronósticos entre todos los tumores cutáneos. Es muy agresivo y tiene altas tasas de recidiva local. La tasa de supervivencia a los 5 años oscila entre el 12% y el 34% según la mayoría de los estudios,1,2 pero puede llegar al 62%.3 A diferencia de otros sarcomas, el grado de diferenciación no se ha asociado con el pronóstico del angiosarcoma cutáneo.4
La forma clásica del angiosarcoma cutáneo es una lesión edematosa mal definida con una presentación clínica mayoritariamente indolente en sus fases iniciales. Aparece en la cara o en el cuero cabelludo de pacientes de edad avanzada (angiosarcoma de Wilson-Jones) y representa aproximadamente el 50% de todos los angiosarcomas cutáneos primarios.5-8 Otras dos formas típicas de angiosarcoma son el síndrome de Stewart-Treves, que se desarrolla en áreas de linfedema de larga duración y es particularmente frecuente en mujeres que se han sometido a una mastectomía radical,9-11 y el angiosarcoma postradiación, que se desarrolla en áreas de piel irradiada, particularmente en la región pectoral de mujeres con antecedentes de cáncer de mama tratado con radioterapia.12-15
El aspecto histopatológico del angiosarcoma cutáneo varía desde formas relativamente diferenciadas con espacios vasculares reconocibles cubiertos por células endoteliales prominentes con cierta atipia y un patrón infiltrativo que diseca los haces de colágeno hasta formas más sólidas altamente indiferenciadas compuestas por células fusiformes o epitelioides con una atipia y pleomorfismo considerablemente mayores y un mayor número de mitosis. Los espacios vasculares son raros y los tumores a veces pueden simular un carcinoma.
El tratamiento principal para el angiosarcoma cutáneo -y el único que es potencialmente curativo si se consiguen márgenes libres de enfermedad- es la escisión quirúrgica con márgenes amplios seguida de radioterapia local e incluso, en opinión de algunos autores, la radiación de los ganglios linfáticos regionales.6 En la mayoría de los casos, sin embargo, no es fácil conseguir márgenes libres de enfermedad debido a la extensa diseminación subclínica. Además, estos tumores son frecuentemente multifocales. La quimioterapia tiene un papel puramente paliativo en el tratamiento del angiosarcoma cutáneo.
Aunque el angiosarcoma cutáneo es poco frecuente (representa menos del 1% de todos los sarcomas), la mayoría de los casos de angiosarcoma se originan en la piel. Debido a su baja prevalencia, los angiosarcomas cutáneos se incluyen en las series de angiosarcomas viscerales u óseos, que tienen un pronóstico aún más sombrío.1 Por lo tanto, existen pocas series amplias de angiosarcomas cutáneos en la literatura debido a la escasez de casos uniformes a largo plazo.2,4,5,7,16 El manejo del angiosarcoma es, además, a menudo desalentador, sobre todo en los casos de enfermedad avanzada, que tienen un pronóstico muy pobre a pesar del uso de un tratamiento agresivo desde el principio. Así pues, motivados por las dificultades asociadas al manejo del angiosarcoma cutáneo y la escasa literatura disponible, investigamos todos los casos de angiosarcoma cutáneo tratados en el Instituto Valenciano de Oncología (IVO), en Valencia, España, con el objetivo de identificar los factores clínicos, histológicos y relacionados con el tratamiento posiblemente asociados al pronóstico. Para ello, se revisaron las historias clínicas y los hallazgos clínicos en busca de datos exploratorios que pudieran servir de guía para el diagnóstico precoz, ya que los pacientes con enfermedad precoz y tumores pequeños tienen una probabilidad de supervivencia considerablemente mayor.
Material y métodos
Realizamos un estudio observacional retrospectivo de todos los casos de angiosarcoma cutáneo tratados en el IVO entre enero de 2000 y diciembre de 2015. Toda la información recopilada se extrajo de las historias clínicas de los pacientes, del archivo de biopsias del departamento de patología y del archivo fotográfico de nuestro departamento. De los 20 casos identificados inicialmente, 4 tuvieron que ser excluidos: 1 porque no había seguimiento, otro porque no había suficiente material para determinar si el tumor era un hemangioendotelioma o un angiosarcoma, y 2 porque los tumores no eran angiosarcomas primarios. Estos 2 tumores habían sido etiquetados inicialmente como angiosarcomas cutáneos porque todos los cortes histológicos mostraban una afectación cutánea de la mama. Sin embargo, al revisar los bloques, comprobamos que la afectación cutánea era secundaria en ambos casos, y que el tumor primario se localizaba en el parénquima mamario desde donde se extendía hasta la piel suprayacente.
Los criterios de inclusión en el estudio fueron los hallazgos clínicos sugestivos de angiosarcoma cutáneo y la confirmación histológica del diagnóstico con tinción de hematoxilina-eosina de las muestras de biopsia, apoyada por estudios inmunohistoquímicos, que en la mayoría de los casos incluían tinción para CD31, CD34, D240 y Ki-67.
Se estudiaron las siguientes variables para cada paciente: edad, sexo, localización y tamaño del tumor, tipo de angiosarcoma (primario, postradiación, asociado a linfedema), tratamiento (cirugía, radioterapia, quimioterapia), recidiva, metástasis, supervivencia y muerte. En el caso de los tumores postradiación y asociados a linfedema, también se registró el tipo de tumor previo y el número de años transcurridos desde la radioterapia o el linfedema. Las variables histológicas analizadas fueron el estado de los márgenes, el patrón histopatológico (vasoformativo, sólido o mixto), el tipo celular predominante (epitelioide o fusiforme), la presencia de necrosis (sí, no), el nivel de invasión (epidermis, dermis, hipodermis, músculo, hueso), la reacción linfocítica, el patrón infiltrativo y el número de mitosis por 10 campos.
Resultados
Se incluyeron en el estudio dieciséis casos de angiosarcoma cutáneo. Correspondían a 11 mujeres y 5 hombres con edades comprendidas entre 35 y 83 años (media, 67 años; mediana, 71 años). Diez de los casos eran angiosarcomas postradiación (10 casos), 5 eran angiosarcomas de Wilson-Jones y sólo 1 era un angiosarcoma asociado a linfedema. La localización más frecuente fue el tronco (10 casos), seguido de la cabeza y el cuello (5 casos). Las extremidades superiores sólo se vieron afectadas en 1 caso. El tamaño más pequeño del tumor fue de 1 cm y el más grande de 50 cm (media, 10 cm; mediana, 6,5 cm).
Siete de los pacientes tenían antecedentes de cáncer (cáncer de mama en 10 casos y seminoma en 1). Con la excepción de 1 caso de carcinoma lobular invasivo, todos los cánceres de mama eran carcinomas ductales invasivos.
El tiempo medio entre la radioterapia y el desarrollo del angiosarcoma en los 10 casos de angiosarcoma postradiación fue de 8,2 años. Sólo 1 de los casos apareció en los 5 años siguientes a la radioterapia; el resto apareció al menos 5 años después.
Catorce casos fueron tratados quirúrgicamente y en 4 de ellos se utilizó radioterapia adyuvante. Ocho pacientes recibieron quimioterapia y éste fue el primer y único tratamiento en 2 pacientes.
La doxorrubicina y el taxol se utilizaron cada uno en 4 casos, la ifosfamida se utilizó en 3 casos, y el paclitaxel y la dacarbazina se utilizaron en 1 caso cada uno. La respuesta a la quimioterapia fue pobre, y aunque casi todos los pacientes mostraron una respuesta parcial, la enfermedad progresó en todos los casos y los pacientes murieron durante el seguimiento (8/8).
Cinco pacientes tuvieron metástasis a distancia, que afectaron a múltiples sitios en la mayoría de los casos. Los lugares más comunes fueron el pulmón y el hígado.
Diez de los 16 pacientes murieron de angiosarcoma durante el seguimiento. Los otros 6 pacientes están actualmente libres de enfermedad. La duración media del seguimiento fue de 42,5 meses (mediana, 26 meses; rango, 7-188 meses).
Histológicamente, 8 casos tenían un patrón de crecimiento sólido, 4 tenían un patrón vasoformativo y 4 tenían un patrón mixto. El tipo celular predominante fue epitelioide en 14 casos y fusiforme en sólo 2. Se observó necrosis en 6 tumores y el patrón infiltrativo fue subcutáneo en la mayoría de los casos (n=10). Cuatro casos se limitaban a la dermis y sólo 2 afectaban a los planos musculares. Los márgenes quirúrgicos no fueron evaluables en 3 casos. De los casos restantes, 8 tenían márgenes negativos y 5 tenían márgenes positivos. La reacción linfocítica fue leve o moderada en 10 casos, intensa en 2, e inexistente en 4. En los 14 casos con reacción linfocítica, el infiltrado fue peritumoral en 2 casos, intratumoral en 8, y mixto en 2. Hubo una media de 15 mitosis por 10 campos (rango, 0-37 mitosis).
Los resultados clínicos y patológicos más relevantes se resumen en la Tabla 1. Los resultados de la comparación entre supervivientes y no supervivientes se resumen en la Tabla 2.
Selección de resultados clínicos y patológicos de los 16 angiosarcomas cutáneos.a
| Paciente | Edad, año | Sexo | Tipo | Localización | Tamaño, cm | Tiempo desde la Rx, mo | Dosis, Gy | Tumor anterior | Tipo de cáncer de mama |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | F | PR | Mama izquierda | 18 | 60 | 46 | Mama | IDC |
| 2 | 71 | F | ST | Brazo izquierdo | 12 | – | – | Mama | IDC |
| 3 | 51 | F | PR | Región submamaria derecha | 1 | 57 | 50 | Mama | IDC |
| 4 | 76 | F | WJ | Cabeza y cuello | 3 | – | – | No | . |
| 5 | 77 | F | PR | Mama derecha | 1 | 94 | 46 | Mama | IDC |
| 6 | 71 | F | PR | Mama izquierda | 50 | 171 | 48 | Mama | IDC |
| 7 | 48 | M | PR | Pared abdominal | 2 | 96 | 26 | Seminoma | – |
| 8 | 55 | F | PR | Mama izquierda | 10 | 88 | 46 | Mama | ILC |
| 9 | 69 | F | PR | Mama izquierda | 8 | 143 | 46 | Mama | IDC |
| 10 | 76 | M | WJ | Mejilla derecha | 6 | – | – | No | – |
| 11 | 35 | F | PR | Seno derecho | 12 | 66 | 50 | Seno | IDC |
| 12 | 57 | F | PR | Mama derecha | 8 | 108 | 50 | Mama | IDC |
| 13 | 68 | M | WJ | Cabeza y cuello | 2 | – | – | No | – |
| 14 | 80 | M | WJ | Cabeza y cuello | 15 | – | – | No | – |
| 15 | 79 | M | WJ | Cabeza y cuello | 2 | – | – | No | – |
| 16 | 83 | F | PR | Seno izquierdo | 3 | 110 | 50 | Seno | IDC |
| X=67.1 | 11W, 5M | 10 RI, 5 WJ, 1 ST | 9 pecho, 5 cabeza y cuello, 1 abdomen, 1 miembro superior | X=10 | X=100.3 | X=45.8 | 10 mama, 1 seminoma |
10 cánceres de mama: 9 IDC, 1 ILC |
| Paciente | Cirugía | Margen, cm | Tratamiento | Muerte | Patrón HP | Tipo de célula | Necrosis | DoI | Mitosis/mm2 | Supervivencia, mo |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Sí | 0.2 | 13 | Sí | 1 | E | No | 3 | 7 | 24 |
| 2 | No | ns | 3 | Sí | 2 | E | No | 3 | 37 | 8 |
| 3 | Sí | 35 | 1 | No | 1 | E | No | 3 | 2 | 29 |
| 2 | 123 | Sí | 2 | E | No | 3 | 28 | 26 | ||
| 5 | Sí | ns | 1 | Sí | 2 | E | No | 2 | 14 | 8 |
| 6 | No | ns | 3 | Sí | 1 | E | No | 2 | 0 | 19 |
| 7 | Sí | ns | 1 | No | 3 | E | No | 3 | 6 | 187 |
| 8 | Sí | ns | 13 | Sí | 2 | E | Sí | 4 | 6 | 28 |
| 9 | Sí | ns | 13 | Sí | 3 | E | Sí | 3 | 22 | 24 |
| 10 | Sí | ns | 12 | Sí | 2 | E | Sí | 3 | 16 | 7 |
| 11 | Sí | 0.5 | 1 | No | 2 | SC | No | 3 | 18 | 76 |
| 12 | Sí | 0 | 13 | Sí | 3 | E | Sí | 3 | 36 | 26 |
| 13 | Sí | 2 | 12 | No | 3 | E | No | 3 | 5 | 95 |
| 14 | Sí | 2 | 123 | Sí | 2 | E | Sí | 4 | 17 | 24 |
| 15 | Sí | 2 | 1 | No | 2 | E | Sí | 2 | 6 | 53 |
| 16 | Sí | 3 | 1 | No | 1 | SC | No | 2 | 23 | 51 |
| 14 sí 2 no | 1.68 | 14: Qx 4: Rt 8: Qt |
10 sí 6 no |
4 vasof 8 sólido 4 mixto |
14 E, 2 SC | 6 Sí 10 No |
4 dermis 10 hipodermis 2 músculo |
X=15 | X=42.8 |
Los pacientes que murieron se muestran en negrita.
Tratamiento AS, tratamiento del angiosarcoma (1, cirugía ; 2, radioterapia ; 3, quimioterapia ); DoI, profundidad de la invasión (2, dermis; 3, hipodermis; 4, músculo); E, epitelioide; HP, histopatológico (1, vasoformativo ; 2,sólido; 3, mixto); IDC, carcinoma ductal invasivo; ILC, carcinoma lobular invasivo; M, hombre; ns, no especificado; PR, angiosarcoma postradiación; Rx, radiación; SC, células fusiformes; ST, angiosarcoma de Stewart-Treves; W, mujer; WJ, angiosarcoma de Wilson-Jones: X, media.
Comparación de variables entre los supervivientes y los pacientes que murieron de angiosarcoma cutáneo.
| Variable | Vivos (n=6) | Fallecidos (n=10) | |
|---|---|---|---|
| Edad, media, y | 61 | 71 | |
| Mujeres | 3 | 8 | |
| Hombres | 3 | 2 | |
| Postradiación | 4 | 6 | |
| Idiopática | 2 | 3 | |
| Linfedema-asociado | 0 | 1 | |
| Tronco | 4 | 6 | |
| Cabeza y cuello | 2 | 3 | |
| Miembros superiores | 0 | 1 | |
| Talla, cm | 3.6 | 13,1 | |
| Tiempo desde la radioterapia, mo | 82,25 | 110.6 | |
| Dosis de radiación, Gy | 44 | 47 | |
| Mama | 3 | 7 | |
| Seminoma | 1 | ||
| Cirugía | 6 | 8 | |
| Radioterapia | 1 | 3 | |
| Quimioterapia | 0 | 0 | 8 |
| Vasoformativo | 2 | 2 | |
| Sólido | 2 | 6 | |
| Mixto | 2 | 2 | |
| Necrosis | 1 | 5 | |
| Dermis | 2 | 2 | |
| Subcutánea | 4 | 6 | |
| Músculo | 0 | 2 | |
| Mitosis | 10 | 183 | |
| Supervivencia, mo | 81,8 | 19.4 | |
Discusión
El angiosarcoma cutáneo es un tumor muy infrecuente, lo que se pone de manifiesto por el hecho de que sólo hemos podido recopilar datos sobre 16 tumores diagnosticados durante un período de 14 años en un hospital oncológico. En general, el angiosarcoma cutáneo es algo más frecuente en los hombres de edad avanzada. Esto se debe a que la forma más común de angiosarcoma cutáneo en la población general es el angiosarcoma primario de cabeza y cuello (también conocido como angiosarcoma idiopático o de Wilson-Jones) (Fig. 1), que tiende a afectar a los hombres de edad avanzada.5,7,17 El angiosarcoma por radiación (Fig. 2) es ahora la segunda forma más común de angiosarcoma cutáneo debido al aumento del uso de la radioterapia frente a la mastectomía radical para tratar el cáncer de mama.2,13,18 Este cambio también ha llevado a una reducción de la frecuencia del angiosarcoma asociado al linfedema, que es actualmente la forma menos común de este tumor. La prevalencia de las diferentes formas de angiosarcoma cutáneo en nuestra serie no coincide con los informes en la literatura, ya que nuestro hospital trata a un gran número de mujeres con cáncer de mama, lo que explica que la forma más común de angiosarcoma cutáneo en nuestro hospital fuera el angiosarcoma postradiación. Este predominio de casos de cáncer de mama también explica que 11 de los 16 pacientes fueran mujeres. Sólo hubo 1 caso de angiosarcoma asociado a linfedema (Fig. 3), lo que coincide con las tasas de prevalencia comunicadas en otros lugares. La paciente tenía un linfedema crónico en el brazo izquierdo secundario a la disección de los ganglios linfáticos axilares realizada como parte del tratamiento del cáncer de mama 22 años antes.
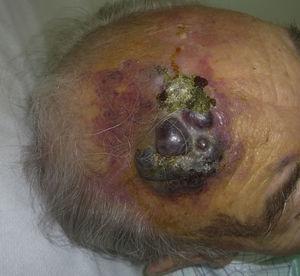
Placa rojiza-violácea con áreas nodulares en la frente de un varón de edad avanzada.
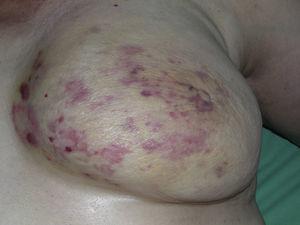
Múltiples máculas y manchas eritematosas con varias pápulas rojizas sobre piel amarillenta en una mama irradiada por cáncer.
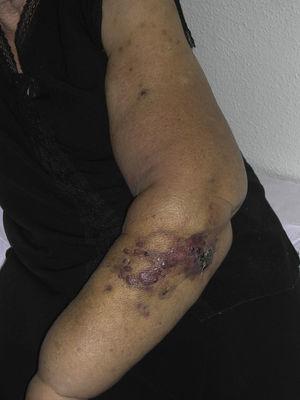
Pápulas y nódulos rojizos-violáceos con áreas similares a hematomas en un brazo con linfedema secundario a una cirugía de cáncer de mama.
Con la excepción de 1 mujer que tenía un angiosarcoma idiopático o de Wilson-Jones, el resto de las mujeres de nuestra serie (n=10) tenían antecedentes de cáncer de mama. Nueve de estos cánceres eran carcinomas ductales invasivos. Dado que el 80% de los cánceres de mama en la población general son carcinomas ductales invasivos y sólo el 10% son carcinomas lobulillares, la alta prevalencia de carcinoma ductal invasivo en nuestro estudio es probablemente un reflejo de la alta prevalencia de este cáncer en la población general, más que de una asociación particular entre el angiosarcoma cutáneo postradiación y el carcinoma ductal invasivo, como parecen sugerir nuestros resultados. En otras palabras, las proporciones observadas en nuestra serie están en consonancia con las tasas descritas para diferentes cánceres de mama en la población general. Hay 2 casos de angiosarcoma de mama incluidos inicialmente en nuestro estudio que merecen ser mencionados. Se ha descrito que el angiosarcoma de mama tiende a ser cutáneo cuando es inducido por radiación y parenquimatoso cuando no lo es.19 En nuestra serie, de los 18 casos de angiosarcoma cutáneo identificados inicialmente, hubo 2 casos de angiosarcoma de mama no inducidos por radioterapia. Al revisar estos casos, comprobamos que la localización primaria del tumor era el parénquima mamario y no la piel. Por lo tanto, los tumores se excluyeron del estudio, ya que eran secundarios y no primarios. Los casos restantes eran todos angiosarcomas mamarios primarios inducidos por la radiación, en consonancia con los informes de la literatura.
La localización más común del angiosarcoma en nuestra serie fue la mama (10 casos) y no la cara o el cuero cabelludo, como cabría esperar. El único caso que afectó a las extremidades fue un angiosarcoma asociado a un linfedema. De nuevo, el hecho de que la localización tumoral más frecuente fuera la mama se explicaría por el predominio de los tumores inducidos por la radiación en nuestra serie.
Aunque el período de latencia entre la exposición a la radioterapia y el desarrollo del angiosarcoma es muy variable según los informes de la literatura (3-50 años), tiende a ser más largo (una media de 25 años) cuando la enfermedad tratada por la radiación es benigna. Los periodos de latencia reportados para las enfermedades malignas son más cortos (alrededor de 10-15 años), excepto en el caso del angiosarcoma de mama, para el que se ha descrito una media de aproximadamente 5 años.12 La razón de este periodo más corto en el caso del angiosarcoma de mama no está clara, aunque se han propuesto varias teorías, como el gran volumen de piel irradiada, la presencia de linfedema asociado, factores posiblemente intrínsecos a la mama y un posible efecto sinérgico con la quimioterapia.13,14 En nuestros 10 casos postradiación, el angiosarcoma se diagnosticó tras una media de 8,2 años, y en el 90% de los casos habían transcurrido al menos 5 años. No se ha descrito ninguna asociación entre el período de latencia y el pronóstico del angiosarcoma. En nuestra serie, el tiempo medio transcurrido desde la radioterapia hasta el desarrollo del angiosarcoma fue algo mayor en los pacientes que fallecieron (110,6 meses) que en los que sobrevivieron (82,25 meses). En el único caso de síndrome de Stewart-Treves, el paciente había sido sometido a una disección de ganglios linfáticos 22 años antes. Los periodos de latencia de los angiosarcomas asociados a linfedema son muy variables, con un rango de 1 a 30 años y una media de 10. El síndrome de Stewart-Treves representa el 90% de todos los casos de angiosarcoma asociado a linfedema.9,11 Sin embargo, el angiosarcoma también puede surgir en otras formas de linfedema, como el linfedema congénito, el linfedema filarial y el linfedema secundario a la disección de ganglios linfáticos en otras partes del cuerpo.10
El tamaño del tumor es actualmente el marcador pronóstico más aceptado para el angiosarcoma cutáneo, y se ha informado con frecuencia que los angiosarcomas que miden 5 cm o más tienen un peor pronóstico que los tumores más pequeños.5,17 En nuestra serie, observamos diferencias en el tamaño medio del tumor entre los supervivientes y los no supervivientes. Los que sobrevivieron tenían un tamaño medio del tumor de 3,6 cm, que era más de 3 veces menor que el tamaño medio (13,1 cm) en el grupo de pacientes que murieron. Nuestros resultados apoyan, por tanto, la creencia de que los tumores de mayor tamaño se asocian a peores resultados en el angiosarcoma, y destacan la importancia de diagnosticar el tumor cuando comienza como una lesión parecida a un hematoma, aunque esto es particularmente difícil en los angiosarcomas del cuero cabelludo y aún más cuando el paciente todavía tiene pelo (Fig. 4). El angiosarcoma de mama postradicional es más fácil de diagnosticar, ya que cualquier lesión persistente en esta zona con apariencia vascular o de hematoma debe ser biopsiada. Una pista diagnóstica potencialmente útil es un halo amarillento (correspondiente a la hemosiderina) alrededor de la lesión sospechosa (Fig. 5), ya que nunca hemos observado este signo en proliferaciones vasculares benignas en la piel irradiada. El angiosarcoma post-radiación y las proliferaciones vasculares atípicas en la piel irradiada deben distinguirse histológicamente, pero las características clínicas pueden ayudar. Las proliferaciones vasculares atípicas tienden a ser mucho más pequeñas que los angiosarcomas y también suelen tener un periodo de latencia más corto.20 A diferencia de los angiosarcomas, estas proliferaciones se limitan a la dermis superficial y media y no invaden el tejido subcutáneo. Además, la histología no muestra la atipia nuclear característica observada en el angiosarcoma, ni las múltiples capas de células endoteliales o las figuras mitóticas. No obstante, en ocasiones puede ser difícil distinguir entre las dos entidades y se ha informado de la coexistencia de las dos lesiones en la misma mama irradiada. Incluso se han dado casos de proliferaciones vasculares atípicas inducidas por la radiación que progresan a angiosarcoma.21 En casos muy complicados, puede ser útil buscar la sobreexpresión del gen MYC si se realiza un estudio inmunohistoquímico, o la amplificación si se utiliza la hibridación in situ fluorescente. La amplificación de MYC es bastante común en los angiosarcomas secundarios, pero no parece ocurrir en las proliferaciones vasculares atípicas en la piel irradiada.22
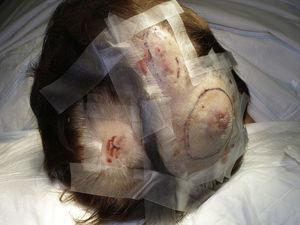
Nódulos eritematosos multifocales en el cuero cabelludo.
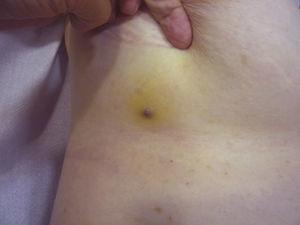
Pápula rojiza-violácea que mide menos de 1 cm rodeada por una zona amarillenta de la piel.
El otro marcador pronóstico más aceptado en el angiosarcoma es la edad. En nuestra serie, los pacientes que sobrevivieron eran en promedio más jóvenes que los que murieron (62 frente a 71 años). No obstante, 10 de los 16 pacientes de nuestra serie habían fallecido al final del estudio, lo que corrobora el mal pronóstico generalmente asociado al angiosarcoma.
A diferencia del caso de otros sarcomas, el American Joint Committee on Cancer no considera que el grado histológico sea un marcador pronóstico relevante en el angiosarcoma. Sin embargo, varios autores han sugerido que ciertas características histopatológicas podrían influir en el pronóstico. Algunos estudios recientes, por ejemplo, han afirmado que un patrón sólido predominante podría ser un marcador de pronóstico relativamente bueno en el angiosarcoma de cabeza y cuello.3,16 Este no fue el caso en nuestra serie, ya que 6 de los 10 pacientes que fallecieron tenían este patrón en comparación con sólo 2 de los 6 pacientes que seguían vivos al final del estudio (Figs. 6 y 7).
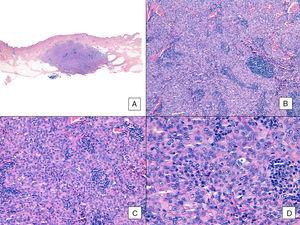
Angiosarcoma con patrón sólido predominante. A, La imagen panorámica muestra la invasión de la dermis reticular media y profunda y de la hipodermis (hematoxilina-eosina, aumento original ×10). B, Tumor densamente celular que ha destruido estructuras preexistentes y se acompaña de infiltrados linfoides nodulares (hematoxilina-eosina, aumento original ×100). C y D, Vista detallada que muestra un predominio de células epitelioides acompañadas de un infiltrado linfocítico (hematoxilina-eosina ×200 y ×400, respectivamente).
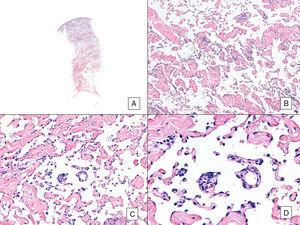
Angiosarcoma con un patrón vasoformativo predominante. A, Imagen panorámica que muestra la infiltración de la dermis hasta la hipodermis (hematoxilina-eosina, aumento original ×10). B, Espacios vasculares neoplásicos con marcada disección del colágeno (hematoxilina-eosina, aumento original ×100). C y D, Vasos no neoplásicos disecados por células endoteliales neoplásicas, que quedan «flotando» en la dermis (signo del promontorio) (hematoxilina-eosina, aumento original ×200 y ×400, respectivamente).
Otros factores histológicos potencialmente de alto riesgo propuestos en la literatura son la presencia de necrosis, una morfología epitelioide y una mayor profundidad de invasión.5,7 Nuestros resultados apoyan estos posibles marcadores, ya que las 3 variables fueron más frecuentes en el grupo de no supervivientes. La necrosis se observó en 5 de los 10 pacientes que fallecieron y en sólo 1 de los 6 pacientes que sobrevivieron. Asimismo, los únicos 2 pacientes con un patrón de células fusiformes predominante seguían vivos al final del estudio. En cambio, ninguno de los pacientes que fallecieron presentaba este patrón. Por último, los únicos 2 pacientes con afectación de los planos musculares murieron. El número medio de mitosis también fue mayor en el grupo de no supervivientes (18,3 frente a 10).
El tratamiento de elección del angiosarcoma cutáneo es la escisión quirúrgica con márgenes amplios seguida de radioterapia.6 La frecuente naturaleza multifocal del angiosarcoma y su propensión a desarrollar una diseminación subclínica a menudo complican la obtención de márgenes claros. Además, no existe un acuerdo universal sobre lo que constituye un margen adecuado, y la mayoría de los estudios proporcionan información imprecisa, como la escisión con «márgenes amplios». En nuestra serie, la cirugía fue el tratamiento de elección en 14 de los 16 pacientes. Se utilizaron márgenes de 3 cm cuando fue técnicamente posible, y en los demás casos se intentó la extirpación utilizando márgenes de 2 cm. Los 14 pacientes fueron sometidos a cirugía radical. Los otros 2 no fueron considerados candidatos a la cirugía y recibieron quimioterapia paliativa. Murieron durante el seguimiento. La radioterapia adyuvante al lecho quirúrgico se utilizó sólo en 4 pacientes. Este bajo uso de la radioterapia en el tratamiento del angiosarcoma en nuestra serie puede explicarse probablemente por el gran número de casos de angiosarcomas postradiación en nuestra serie. En otras palabras, probablemente influyó un cierto nivel de resistencia al uso de la radiación en estos casos. Sin embargo, el uso de la radioterapia en los angiosarcomas postradiación encuentra apoyo en la literatura, e incluso a veces se administra como monoterapia, sin cirugía.23,24 La quimioterapia se utilizó en la mitad de los pacientes. Como ya se ha mencionado, tuvo un papel meramente paliativo y se asoció a un mal resultado en todos los casos. Aunque la monoterapia con paclitaxel no fue el régimen de quimioterapia más utilizado en nuestra serie (ya que los casos se diagnosticaron hace tiempo), este régimen se utiliza actualmente como opción de primera línea en la mayoría de los casos. A pesar de las expectativas iniciales generadas, los fármacos angiogénicos (sunitinib, sorafenib, bevacizumab, talidomida) no se utilizan en este entorno debido a sus decepcionantes resultados.
A pesar del reducido número de casos de nuestra serie de angiosarcoma cutáneo, que nos impidió realizar análisis estadísticos, observamos que tanto el mayor tamaño del tumor como la mayor edad se asociaron a un peor pronóstico. También se observó una asociación menos evidente entre el mal pronóstico y las siguientes características histológicas: presencia de necrosis, predominio de células epitelioides, invasión de las capas más profundas y mayor número de mitosis.
Divulgaciones éticasProtección de seres humanos y animales
Los autores declaran que no se han realizado pruebas en seres humanos ni en animales para la realización de este estudio.
Confidencialidad de los datos
Los autores declaran que han seguido el protocolo de su hospital sobre la publicación de datos relativos a los pacientes.
Derecho a la intimidad y consentimiento informado
Los autores declaran que en este artículo no aparecen datos privados de pacientes.
Conflictos de intereses
Los autores declaran que no tienen conflictos de intereses.