Das kutane Angiosarkom hat eine der schlechtesten Prognosen unter allen Hauttumoren. Es ist sehr aggressiv und hat hohe Lokalrezidivraten. Die 5-Jahres-Überlebensrate liegt den meisten Studien zufolge zwischen 12 % und 34 %,1,2 kann aber auch bis zu 62 % betragen.3 Im Gegensatz zu anderen Sarkomen wurde der Differenzierungsgrad beim kutanen Angiosarkom nicht mit der Prognose in Verbindung gebracht.4
Die klassische Form des kutanen Angiosarkoms ist eine schlecht definierte, blaue, ödematöse Läsion mit einem weitgehend indolenten klinischen Bild in der Frühphase. Es tritt im Gesicht oder auf der Kopfhaut älterer Patienten auf (Wilson-Jones-Angiosarkom) und macht etwa 50 % aller primären kutanen Angiosarkome aus.5-8 Zwei weitere typische Formen des Angiosarkoms sind das Stewart-Treves-Syndrom, das sich in Bereichen langjähriger Lymphödeme entwickelt und besonders häufig bei Frauen auftritt, die sich einer radikalen Mastektomie unterzogen haben9-11 , und das Angiosarkom nach Bestrahlung, das sich in Bereichen bestrahlter Haut entwickelt, insbesondere im Brustbereich von Frauen, die in der Vorgeschichte Brustkrebs hatten und mit Strahlentherapie behandelt wurden.12-15
Das histopathologische Erscheinungsbild des kutanen Angiosarkoms variiert von relativ differenzierten Formen mit erkennbaren Gefäßräumen, die von prominenten Endothelzellen mit einer gewissen Atypie und einem infiltrativen, die Kollagenbündel durchdringenden Muster bedeckt sind, bis hin zu solideren, hochgradig undifferenzierten Formen, die aus spindelförmigen oder epitheloiden Zellen mit erheblich größerer Atypie und Pleomorphie sowie einer höheren Zahl von Mitosen bestehen. Gefäßräume sind selten, und die Tumoren können manchmal ein Karzinom imitieren.
Die Hauptbehandlung für kutane Angiosarkome – und die einzige, die potenziell kurativ ist, wenn krankheitsfreie Ränder erreicht werden – ist die chirurgische Exzision mit breiten Rändern, gefolgt von einer lokalen Strahlentherapie und nach Meinung einiger Autoren sogar einer Bestrahlung regionaler Lymphknoten.6 In den meisten Fällen ist es jedoch nicht einfach, krankheitsfreie Ränder zu erreichen, da eine umfangreiche subklinische Ausbreitung vorliegt. Darüber hinaus sind diese Tumoren häufig multifokal. Die Chemotherapie spielt bei der Behandlung des kutanen Angiosarkoms eine rein palliative Rolle.
Obwohl das kutane Angiosarkom selten ist (es macht weniger als 1 % aller Sarkome aus), haben die meisten Angiosarkome ihren Ursprung in der Haut. Aufgrund ihrer geringen Prävalenz werden kutane Angiosarkome in Serien von viszeralen oder knöchernen Angiosarkomen eingeschlossen, die eine noch schlechtere Prognose haben.1 Es gibt daher nur wenige große Serien von kutanen Angiosarkomen in der Literatur, da es an langfristigen, einheitlichen Fällen mangelt.2,4,5,7,16 Das Management von Angiosarkomen ist zudem oft entmutigend, vor allem bei fortgeschrittener Erkrankung, die trotz aggressiver Behandlung von Anfang an eine sehr schlechte Prognose hat. Motiviert durch die Schwierigkeiten, die mit der Behandlung von kutanen Angiosarkomen verbunden sind, und die geringe verfügbare Literatur untersuchten wir alle Fälle von kutanen Angiosarkomen, die am Instituto Valenciano de Oncología (IVO) in Valencia, Spanien, behandelt wurden, mit dem Ziel, klinische, histologische und behandlungsbezogene Faktoren zu identifizieren, die möglicherweise mit der Prognose in Zusammenhang stehen. Dazu überprüften wir die Krankenakten und klinischen Befunde auf der Suche nach explorativen Daten, die als Leitfaden für eine frühzeitige Diagnose dienen könnten, da Patienten mit frühzeitiger Erkrankung und kleinen Tumoren eine deutlich bessere Überlebenswahrscheinlichkeit haben.
Material und Methoden
Wir führten eine retrospektive Beobachtungsstudie aller Fälle von kutanen Angiosarkomen durch, die zwischen Januar 2000 und Dezember 2015 im IVO behandelt wurden. Alle gesammelten Informationen wurden aus den Krankenakten der Patienten, dem Biopsiearchiv der Pathologie und dem Fotoarchiv unserer Abteilung entnommen. Von den 20 ursprünglich identifizierten Fällen mussten 4 ausgeschlossen werden: Einer, weil es keine Nachuntersuchung gab, ein anderer, weil nicht genügend Material vorhanden war, um festzustellen, ob es sich um ein Hämangioendotheliom oder ein Angiosarkom handelte, und 2, weil es sich bei den Tumoren nicht um primäre Angiosarkome handelte. Diese beiden Tumore waren ursprünglich als kutane Angiosarkome eingestuft worden, da alle histologischen Präparate eine kutane Beteiligung der Brust zeigten. Bei der Überprüfung der Blöcke stellten wir jedoch fest, dass die kutane Beteiligung in beiden Fällen sekundär war und der Primärtumor im Brustparenchym lokalisiert war, von wo aus er sich auf die darüber liegende Haut ausdehnte.
Die Einschlusskriterien für die Studie waren klinische Befunde, die auf ein kutanes Angiosarkom hindeuten, und die histologische Bestätigung der Diagnose durch Hämatoxylin-Eosin-Färbung von Biopsieproben, unterstützt durch immunhistochemische Untersuchungen, die in den meisten Fällen CD31-, CD34-, D240- und Ki-67-Färbung umfassten.
Die folgenden Variablen wurden für jeden Patienten untersucht: Alter, Geschlecht, Lage und Größe des Tumors, Art des Angiosarkoms (primär, nach der Bestrahlung, Lymphödem-assoziiert), Behandlung (Operation, Strahlentherapie, Chemotherapie), Rezidiv, Metastasierung, Überleben und Tod. Bei Tumoren nach Bestrahlung und Lymphödem-assoziierten Tumoren wurden auch die Art des vorherigen Tumors und die Anzahl der Jahre seit der Bestrahlungstherapie oder dem Lymphödem erfasst. Die analysierten histologischen Variablen waren der Randstatus, das histopathologische Muster (vasoformativ, solide oder gemischt), der vorherrschende Zelltyp (epitheloid oder spindelzellig), das Vorhandensein von Nekrosen (ja, nein), der Grad der Invasion (Epidermis, Dermis, Hypodermis, Muskel, Knochen), die lymphozytäre Reaktion, das Infiltrationsmuster und die Anzahl der Mitosen pro 10 Felder.
Ergebnisse
Sechzehn Fälle von kutanen Angiosarkomen wurden in die Studie aufgenommen. Es handelte sich um 11 Frauen und 5 Männer im Alter zwischen 35 und 83 Jahren (Mittelwert, 67 Jahre; Median, 71 Jahre). Bei zehn der Fälle handelte es sich um ein Angiosarkom nach Bestrahlung (10 Fälle), bei fünf um ein Wilson-Jones-Angiosarkom und nur bei einem um ein Lymphödem-assoziiertes Angiosarkom. Die häufigste Lokalisation war der Rumpf (10 Fälle), gefolgt von Kopf und Hals (5 Fälle). Die oberen Extremitäten waren nur in 1 Fall betroffen. Die kleinste Tumorgröße betrug 1 cm und die größte 50 cm (Mittelwert, 10 cm; Median, 6,5 cm).
Elf der Patienten hatten eine Krebsvorgeschichte (Brustkrebs in 10 Fällen und Seminom in 1 Fall). Mit Ausnahme von 1 Fall von invasivem lobulärem Karzinom waren alle Brustkrebse invasive duktale Karzinome.
Die mittlere Zeit zwischen der Strahlentherapie und der Entwicklung eines Angiosarkoms in den 10 Fällen von Angiosarkomen nach der Bestrahlung betrug 8,2 Jahre. Nur in einem der Fälle trat das Angiosarkom innerhalb von 5 Jahren nach der Strahlentherapie auf, in den übrigen Fällen mindestens 5 Jahre später.
In 14 Fällen wurde eine chirurgische Behandlung durchgeführt, in 4 Fällen eine adjuvante Strahlentherapie. Acht Patienten erhielten eine Chemotherapie, und bei 2 Patienten war dies die erste und einzige Behandlung.
Doxorubicin und Taxol wurden jeweils in 4 Fällen eingesetzt, Ifosfamid in 3 Fällen und Paclitaxel und Dacarbazin in je einem Fall. Das Ansprechen auf die Chemotherapie war schlecht, und obwohl fast alle Patienten ein teilweises Ansprechen zeigten, schritt die Krankheit in allen Fällen fort, und die Patienten starben während der Nachbeobachtung (8/8).
Fünf Patienten hatten Fernmetastasen, die in den meisten Fällen mehrere Stellen betrafen. Die häufigsten Lokalisationen waren die Lunge und die Leber.
Zehn der 16 Patienten starben während der Nachbeobachtung an einem Angiosarkom. Die anderen 6 Patienten sind derzeit frei von der Krankheit. Die durchschnittliche Nachbeobachtungszeit betrug 42,5 Monate (Median, 26 Monate; Bereich, 7-188 Monate).
Histologisch gesehen hatten 8 Fälle ein solides Wachstumsmuster, 4 ein vasoformatives Muster und 4 ein gemischtes Muster. Der vorherrschende Zelltyp war in 14 Fällen epitheloid und in nur 2 Fällen spindelförmig. 6 Tumore wiesen eine Nekrose auf, und das infiltrative Muster war in den meisten Fällen subkutan (n=10). Vier Fälle beschränkten sich auf die Dermis und nur 2 betrafen die Muskelebenen. In 3 Fällen waren die Operationsränder nicht auswertbar. Von den übrigen Fällen hatten 8 negative und 5 positive Ränder. Die lymphozytäre Reaktion war in 10 Fällen leicht oder mäßig, in 2 Fällen intensiv und in 4 Fällen nicht vorhanden. In den 14 Fällen mit einer lymphozytären Reaktion war das Infiltrat in 2 Fällen peritumoral, in 8 Fällen intratumoral und in 2 Fällen gemischt. Es gab einen Mittelwert von 15 Mitosen pro 10 Felder (Bereich, 0-37 Mitosen).
Die wichtigsten klinischen und pathologischen Ergebnisse sind in Tabelle 1 zusammengefasst. Die Ergebnisse des Vergleichs zwischen Überlebenden und Nicht-Überlebenden sind in Tabelle 2 zusammengefasst.
Auswahl der klinischen und pathologischen Ergebnisse für die 16 kutanen Angiosarkome.a
| Patient | Alter, j | Geschlecht | Typ | Lokalisation | Größe, cm | Zeit seit Rx, mo | Dosis, Gy | Vorheriger Tumor | Brustkrebs Typ |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | F | PR | Linke Brust | 18 | 60 | 46 | Brust | IDC |
| 2 | 71 | F | ST | Linker Arm | 12 | – | – | Brust | IDC |
| 3 | 51 | F | PR | Rechte submammäre Region | 1 | 57 | 50 | Brust | IDC |
| 4 | 76 | F | WJ | Kopf und Hals | 3 | – | – | Nein | . |
| 5 | 77 | F | PR | Rechte Brust | 1 | 94 | 46 | Brust | IDC |
| 6 | 71 | F | PR | Linke Brust | 50 | 171 | 48 | Brust | IDC |
| 7 | 48 | M | PR | Bauchdecke | 2 | 96 | 26 | Seminom | – |
| 8 | 55 | F | PR | Linke Brust | 10 | 88 | 46 | Brust | ILC |
| 9 | 69 | F | PR | Linke Brust | 8 | 143 | 46 | Brust | IDC |
| 10 | 76 | M | WJ | Rechte Wange | 6 | – | – | Nein | – |
| 11 | 35 | F | PR | Rechte Brust | 12 | 66 | 50 | Brust | IDC |
| 12 | 57 | F | PR | Rechte Brust | 8 | 108 | 50 | Brust | IDC |
| 13 | 68 | M | WJ | Kopf und Hals | 2 | – | – | Nein | – |
| 14 | 80 | M | WJ | Kopf und Hals | 15 | – | – | Nein | – |
| 15 | 79 | M | WJ | Kopf und Hals | 2 | – | – | Nein | – |
| 16 | 83 | F | PR | Linke Brust | 3 | 110 | 50 | Brust | IDC |
| X=67.1 | 11W, 5M | 10 RI, 5 WJ, 1 ST | 9 Brust, 5 Kopf und Hals, 1 Abdomen, 1 obere Extremität | X=10 | X=100.3 | X=45.8 | 10 Brust, 1 Seminom |
10 Mammakarzinome: 9 IDC, 1 ILC |
| Patientin | Chirurgie | Margin, cm | Als Behandlung | Tod | HP-Muster | Zelltyp | Nekrose | DoI | Mitosen/mm2 | Überleben, mo |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ja | 0.2 | 13 | Ja | 1 | E | Nein | 3 | 7 | 24 |
| 2 | Nein | ns | 3 | Ja | 2 | E | Nein | 3 | 37 | 8 |
| 3 | Ja | 3.5 | 1 | Nein | 1 | E | Nein | 3 | 2 | 29 |
| 4 | Ja | 2 | 123 | Ja | 2 | E | Nein | 3 | 28 | 26 |
| 5 | Ja | ns | 1 | Ja | 2 | E | Nein | 2 | 14 | 8 |
| 6 | Nein | ns | 3 | Ja | 1 | E | Nein | 2 | 0 | 19 |
| 7 | Ja | ns | 1 | Nein | 3 | E | Nein | 3 | 6 | 187 |
| 8 | Ja | ns | 13 | Ja | 2 | E | Ja | 4 | 6 | 28 |
| 9 | Ja | ns | 13 | Ja | 3 | E | Ja | 3 | 22 | 24 |
| 10 | Ja | ns | 12 | Ja | 2 | E | Ja | 3 | 16 | 7 |
| 11 | Ja | 0.5 | 1 | Nein | 2 | SC | Nein | 3 | 18 | 76 |
| 12 | Ja | 0 | 13 | Ja | 3 | E | Ja | 3 | 36 | 26 |
| 13 | Ja | 2 | 12 | Nein | 3 | E | Nein | 3 | 5 | 95 |
| 14 | Ja | 2 | 123 | Ja | 2 | E | Ja | 4 | 17 | 24 |
| 15 | Ja | 2 | 1 | Nein | 2 | E | Ja | 2 | 6 | 53 |
| 16 | Ja | 3 | 1 | Nein | 1 | SC | Nein | 2 | 23 | 51 |
| 14 ja, 2 nein | 1.68 | 14: Qx 4: Rt 8: Qt |
10 ja 6 nein |
4 vasof 8 fest 4 gemischt |
14 E, 2 SC | 6 ja 10 nein |
4 Dermis 10 Hypodermis 2 Muskel |
X=15 | X=42.8 |
Die Patienten, die gestorben sind, sind fett gedruckt.
AS-Behandlung, Angiosarkom-Behandlung (1, Operation; 2, Strahlentherapie; 3, Chemotherapie); DoI, Invasionstiefe (2, Dermis; 3, Hypodermis; 4, Muskel); E, epithelioid; HP, histopathologisch (1, vasoformativ; 2, solide; 3, gemischt); IDC, invasives duktales Karzinom; ILC, invasives lobuläres Karzinom; M, Mann; ns, keine Angabe; PR, Angiosarkom nach Bestrahlung; Rx, Bestrahlung; SC, spindelzellig; ST, Stewart-Treves-Angiosarkom; W, Frau; WJ, Wilson-Jones-Angiosarkom: X, Mittelwert.
Vergleich der Variablen zwischen Überlebenden und Patienten, die an einem kutanen Angiosarkom gestorben sind.
| Variable | Überlebende (n=6) | Verstorbene (n=10) |
|---|---|---|
| Alter, Durchschnitt, y | 61 | 71 |
| Frauen | 3 | 8 |
| Männer | 3 | 2 |
| Postradiation | 4 | 6 |
| Idiopathisch | 2 | 3 |
| Lymphödem-assoziiert | 0 | 1 |
| Rumpf | 4 | 6 |
| Kopf und Hals | 2 | 3 |
| Obere Gliedmaßen | 0 | 1 |
| Größe, cm | 3.6 | 13.1 |
| Zeit seit Strahlentherapie, mo | 82.25 | 110.6 |
| Strahlendosis, Gy | 44 | 47 |
| Brust | 3 | 7 |
| Seminom | 1 | |
| Chirurgie | 6 | 8 |
| Strahlentherapie | 1 | 3 |
| Chemotherapie | 0 | 8 |
| Vasoformativ | 2 | 2 |
| Fest | 2 | 6 |
| Gemischt | 2 | 2 |
| Nekrose | 1 | 5 |
| Dermis | 2 | 2 |
| Subkutane | 4 | 6 |
| Muskeln | 0 | 2 |
| Mitosen | 10 | 18.3 |
| Überleben, mo | 81,8 | 19.4 |
Diskussion
Das kutane Angiosarkom ist ein sehr seltener Tumor, was durch die Tatsache belegt wird, dass wir nur Daten über 16 Tumore sammeln konnten, die über einen Zeitraum von 14 Jahren in einem onkologischen Krankenhaus diagnostiziert wurden. Insgesamt ist das kutane Angiosarkom bei älteren Männern etwas häufiger anzutreffen. Dies liegt daran, dass die häufigste Form des kutanen Angiosarkoms in der Allgemeinbevölkerung das primäre Angiosarkom im Kopf- und Halsbereich ist (auch bekannt als idiopathisches oder Wilson-Jones-Angiosarkom) (Abb. 1), das tendenziell ältere Männer betrifft.5,7,17 Das Angiosarkom nach Bestrahlung (Abb. 2) ist heute die zweithäufigste Form des kutanen Angiosarkoms, da bei der Behandlung von Brustkrebs vermehrt auf Strahlentherapie statt auf radikale Mastektomie zurückgegriffen wird.2,13,18 Dieser Wandel hat auch zu einem Rückgang der Häufigkeit des Lymphödem-assoziierten Angiosarkoms geführt, das derzeit die seltenste Form dieses Tumors ist. Die Prävalenz der verschiedenen Formen des kutanen Angiosarkoms in unserer Serie stimmt nicht mit den Berichten in der Literatur überein, da in unserem Krankenhaus eine große Anzahl von Frauen mit Brustkrebs behandelt wird, was erklärt, warum die häufigste Form des kutanen Angiosarkoms in unserem Krankenhaus das Angiosarkom nach der Bestrahlung war. Dieses Übergewicht an Brustkrebsfällen erklärt auch, warum 11 der 16 Patienten Frauen waren. Es gab nur 1 Fall eines Lymphödem-assoziierten Angiosarkoms (Abb. 3), was mit den anderswo berichteten Prävalenzraten übereinstimmt. Die Patientin hatte ein chronisches Lymphödem am linken Arm als Folge einer axillären Lymphknotendissektion, die im Rahmen einer Brustkrebsbehandlung 22 Jahre zuvor durchgeführt worden war.
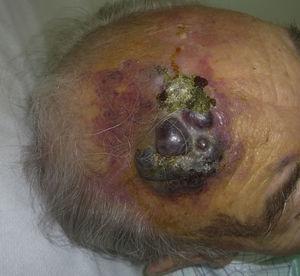
Rötlich-violette Plaque mit knotigen Bereichen auf der Stirn eines älteren Mannes.
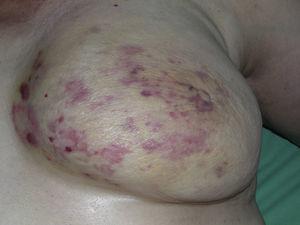
Mehrere Makel und erythematöse Flecken mit mehreren rötlichen Papeln auf gelblicher Haut an einer wegen Krebs bestrahlten Brust.
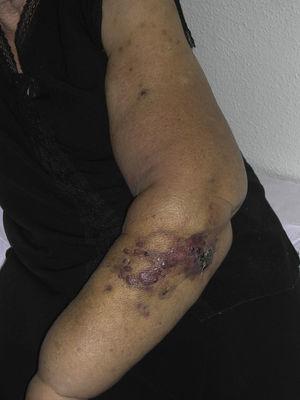
Rötlich-violette Papeln und Knötchen mit blutergussähnlichen Bereichen an einem Arm mit Lymphödem nach einer Brustkrebsoperation.
Mit Ausnahme von 1 Frau, die ein idiopathisches oder Wilson-Jones-Angiosarkom hatte, hatten die übrigen Frauen in unserer Serie (n=10) eine Vorgeschichte von Brustkrebs. Neun dieser Krebsarten waren invasive duktale Karzinome. Da 80 % der Brustkrebsfälle in der Allgemeinbevölkerung invasive duktale Karzinome und nur 10 % lobuläre Karzinome sind, spiegelt die hohe Prävalenz des invasiven duktalen Karzinoms in unserer Studie wahrscheinlich eher die hohe Prävalenz dieses Krebses in der Allgemeinbevölkerung wider als eine besondere Assoziation zwischen dem kutanen Angiosarkom nach der Bestrahlung und dem invasiven duktalen Karzinom, wie unsere Ergebnisse vermuten lassen. Mit anderen Worten, die in unserer Serie beobachteten Proportionen stehen im Einklang mit den für verschiedene Brustkrebsarten in der Allgemeinbevölkerung beschriebenen Raten. Erwähnenswert sind 2 Fälle von Angiosarkomen der Brust, die ursprünglich in unsere Studie aufgenommen wurden. Es wurde berichtet, dass Angiosarkome der Brust tendenziell kutan sind, wenn sie durch Strahlung ausgelöst werden, und parenchymatös, wenn dies nicht der Fall ist.19 In unserer Serie gab es unter den 18 Fällen von kutanen Angiosarkomen, die ursprünglich identifiziert wurden, 2 Fälle von Angiosarkomen der Brust, die nicht durch Strahlentherapie ausgelöst wurden. Bei der Überprüfung dieser Fälle stellten wir fest, dass der primäre Ort des Tumors das Brustparenchym und nicht die Haut war. Die Tumore wurden daher aus der Studie ausgeschlossen, da sie sekundär und nicht primär waren. Bei den verbleibenden Fällen handelte es sich durchweg um primäre Angiosarkome der Brust, die durch Bestrahlung ausgelöst wurden, was mit Berichten in der Literatur übereinstimmt.
Die häufigste Lokalisation für Angiosarkome in unserer Serie war die Brust (10 Fälle) und nicht das Gesicht oder die Kopfhaut, wie zu erwarten wäre. Der einzige Fall, der die Extremitäten betraf, war ein Lymphödem-assoziiertes Angiosarkom. Auch hier erklärt sich die Tatsache, dass die häufigste Tumorstelle die Brust war, durch das Vorherrschen von strahleninduzierten Tumoren in unserer Serie.
Obwohl die Latenzzeit zwischen der Strahlentherapie und der Entwicklung eines Angiosarkoms den Berichten in der Literatur zufolge sehr unterschiedlich ist (3-50 Jahre), ist sie tendenziell länger (im Durchschnitt 25 Jahre), wenn die durch Strahlung behandelte Krankheit gutartig ist. Bei bösartigen Erkrankungen sind die Latenzzeiten kürzer (etwa 10-15 Jahre), außer bei Angiosarkomen der Brust, für die ein Mittelwert von etwa 5 Jahren beschrieben wurde.12 Der Grund für diese kürzere Zeitspanne beim Angiosarkom der Brust ist unklar, obwohl mehrere Theorien vorgeschlagen wurden, darunter das große Volumen der bestrahlten Haut, das Vorhandensein eines begleitenden Lymphödems, möglicherweise brustimmanente Faktoren und ein möglicher Synergieeffekt mit der Chemotherapie.13,14 In unseren 10 Fällen nach der Bestrahlung wurde das Angiosarkom nach durchschnittlich 8,2 Jahren diagnostiziert, und in 90 % der Fälle waren mindestens 5 Jahre vergangen. Ein Zusammenhang zwischen Latenzzeit und Prognose ist für Angiosarkome nicht bekannt. In unserer Serie war die mittlere Zeitspanne zwischen der Strahlentherapie und der Entwicklung eines Angiosarkoms bei den Patienten, die starben (110,6 Monate), etwas länger als bei denen, die überlebten (82,25 Monate). In dem einzigen Fall des Stewart-Treves-Syndroms hatte sich der Patient 22 Jahre zuvor einer Lymphknotendissektion unterzogen. Die gemeldeten Latenzzeiten für Lymphödem-assoziierte Angiosarkome sind sehr unterschiedlich und reichen von 1 bis 30 Jahren mit einem Mittelwert von 10 Jahren. Das Stewart-Treves-Syndrom macht 90 % aller Fälle von Lymphödem-assoziierten Angiosarkomen aus.9,11 Angiosarkome können jedoch auch bei anderen Formen von Lymphödemen auftreten, z. B. bei angeborenen Lymphödemen, Filarien-Lymphödemen und Lymphödemen nach Lymphknotendissektion in anderen Körperregionen.10
Die Tumorgröße ist derzeit der am weitesten akzeptierte prognostische Marker für kutane Angiosarkome, und es wurde häufig berichtet, dass Angiosarkome mit einer Größe von 5 cm oder mehr eine schlechtere Prognose haben als kleinere Tumore.5,17 In unserer Serie beobachteten wir Unterschiede in der mittleren Tumorgröße zwischen Überlebenden und Nicht-Überlebenden. Diejenigen, die überlebten, hatten eine mittlere Tumorgröße von 3,6 cm, was mehr als dreimal kleiner war als die mittlere Größe (13,1 cm) in der Gruppe der Patienten, die starben. Unsere Ergebnisse stützen somit die Annahme, dass größere Tumore bei Angiosarkomen mit schlechteren Ergebnissen einhergehen, und unterstreichen, wie wichtig es ist, den Tumor bereits dann zu diagnostizieren, wenn er als blutergussähnliche Läsion beginnt, obwohl dies bei Angiosarkomen der Kopfhaut besonders schwierig ist, vor allem, wenn der Patient noch Haare hat (Abb. 4). Das Angiosarkom der Brust nach der Bestrahlung ist leichter zu diagnostizieren, da jede persistierende Läsion in diesem Bereich, die ein vaskuläres oder blutergussähnliches Aussehen hat, biopsiert werden sollte. Ein potenziell nützlicher diagnostischer Hinweis ist ein gelblicher Halo (entsprechend Hämosiderin) um die verdächtige Läsion (Abb. 5), da wir dieses Zeichen bei gutartigen Gefäßwucherungen auf bestrahlter Haut noch nie beobachtet haben. Ein Angiosarkom nach Bestrahlung und atypische Gefäßproliferationen auf bestrahlter Haut müssen histologisch abgegrenzt werden, wobei klinische Merkmale hilfreich sein können. Atypische vaskuläre Proliferationen sind in der Regel viel kleiner als Angiosarkome und haben im Allgemeinen auch eine kürzere Latenzzeit.20 Im Gegensatz zu Angiosarkomen sind diese Proliferationen auf die oberflächliche und mittlere Dermis beschränkt und dringen nicht in das subkutane Gewebe ein. Darüber hinaus zeigt die Histologie weder die charakteristische Kernatypie, die bei Angiosarkomen zu beobachten ist, noch die mehrfachen Schichten von Endothelzellen oder mitotische Figuren. Dennoch kann es gelegentlich schwierig sein, zwischen den beiden Entitäten zu unterscheiden, und es gibt Berichte über das gleichzeitige Auftreten der beiden Läsionen in ein und derselben bestrahlten Brust. Es gab sogar Fälle von strahleninduzierten atypischen vaskulären Proliferationen, die sich zu einem Angiosarkom entwickelten.21 In sehr komplizierten Fällen kann es hilfreich sein, nach einer Überexpression des MYC-Gens zu suchen, wenn eine immunhistochemische Untersuchung durchgeführt wird, oder nach einer Amplifikation, wenn eine Fluoreszenz-in-situ-Hybridisierung verwendet wird. Eine MYC-Amplifikation ist bei sekundären Angiosarkomen recht häufig, scheint aber bei atypischen Gefäßwucherungen auf bestrahlter Haut nicht aufzutreten.22
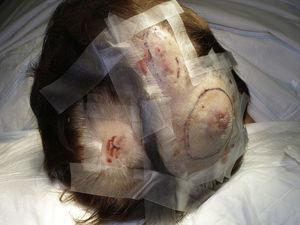
Multifokale erythematöse Knötchen auf der Kopfhaut.
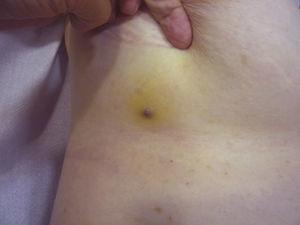
Rötlich-violette Papel von weniger als 1 cm Größe, umgeben von einem gelblichen Hautareal.
Der andere weithin akzeptierte prognostische Marker bei Angiosarkomen ist das Alter. In unserer Serie waren die Patienten, die überlebten, im Durchschnitt jünger als die, die starben (62 versus 71 Jahre). Dennoch waren 10 der 16 Patienten in unserer Serie bis zum Ende der Studie verstorben, was die schlechte Prognose bestätigt, die allgemein mit Angiosarkomen assoziiert wird.
Im Gegensatz zu anderen Sarkomen hält das American Joint Committee on Cancer den histologischen Grad bei Angiosarkomen nicht für einen relevanten Prognosemarker. Mehrere Autoren haben jedoch darauf hingewiesen, dass bestimmte histopathologische Merkmale einen Einfluss auf die Prognose haben könnten. In einigen neueren Studien wurde beispielsweise behauptet, dass ein vorherrschendes solides Muster ein relativ guter Prognosemarker für Angiosarkome im Kopf- und Halsbereich sein könnte.3,16 Dies war in unserer Serie nicht der Fall, da 6 der 10 Patienten, die starben, dieses Muster aufwiesen, verglichen mit nur 2 der 6 Patienten, die am Ende der Studie noch am Leben waren (Abb. 6 und 7).
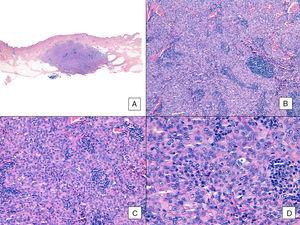
Angiosarkom mit überwiegendem soliden Muster. A, Das Panoramabild zeigt eine Invasion der mittleren und tiefen retikulären Dermis und der Hypodermis (Hämatoxylin-Eosin, Originalvergrößerung ×10). B, dicht zellulärer Tumor, der bereits vorhandene Strukturen zerstört hat und von nodulären lymphatischen Infiltraten begleitet wird (Hämatoxylin-Eosin, Originalvergrößerung ×100). C und D, Detailansicht mit überwiegend epitheloiden Zellen, begleitet von einem lymphoiden Infiltrat (Hämatoxylin-Eosin ×200 bzw. ×400).
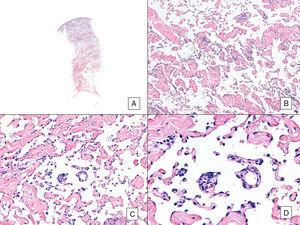
Angiosarkom mit vorherrschendem vasoformativen Muster. A, Panoramaschichtaufnahme mit Infiltration der Dermis bis hinunter zur Hypodermis (Hämatoxylin-Eosin, Originalvergrößerung ×10). B, Neoplastische Gefäßräume mit ausgeprägter Kollagendissektion (Hämatoxylin-Eosin, Originalvergrößerung ×100). C und D, nicht-neoplastische Gefäße, die von neoplastischen Endothelzellen durchtrennt sind, die in der Dermis „schwimmen“ (Promontoriumszeichen) (Hämatoxylin-Eosin, Originalvergrößerung ×200 bzw. ×400).
Weitere in der Literatur vorgeschlagene potenzielle histologische Risikofaktoren sind das Vorhandensein von Nekrosen, eine epithelioide Morphologie und eine größere Tiefe der Invasion.5,7 Unsere Ergebnisse unterstützen diese potenziellen Marker, da alle 3 Variablen in der Gruppe der Nicht-Überlebenden häufiger vorkamen. Eine Nekrose wurde bei 5 der 10 verstorbenen Patienten und nur bei einem der 6 überlebenden Patienten beobachtet. Ebenso waren die einzigen 2 Patienten mit einem vorherrschenden Spindelzellmuster am Ende der Studie noch am Leben. Keiner der Patienten, die starben, wies dagegen dieses Muster auf. Schließlich starben auch die einzigen 2 Patienten mit einer Beteiligung der Muskelebenen. Die durchschnittliche Anzahl der Mitosen war in der Gruppe der Nichtüberlebenden ebenfalls höher (18,3 gegenüber 10).
Die bevorzugte Behandlung des kutanen Angiosarkoms ist die chirurgische Exzision mit breiten Rändern und anschließender Strahlentherapie.6 Die häufige multifokale Natur des Angiosarkoms und seine Neigung zur Entwicklung subklinischer Ausbreitung erschweren oft die Erzielung klarer Ränder. Darüber hinaus gibt es keine allgemeingültige Vereinbarung darüber, was ein angemessener Rand ist, und die meisten Studien geben ungenaue Informationen, wie z. B. die Exzision mit „breiten Rändern“. In unserer Serie war die Operation bei 14 der 16 Patienten die Behandlung der Wahl. Wo es technisch möglich war, wurden 3 cm breite Ränder verwendet, in den anderen Fällen wurde eine Entfernung mit 2 cm breiten Rändern versucht. Die 14 Patienten wurden einer radikalen Operation unterzogen. Die anderen 2 Patienten kamen für eine Operation nicht in Frage und erhielten eine palliative Chemotherapie. Sie starben während der Nachbeobachtung. Eine adjuvante Strahlentherapie am Operationsbett wurde nur bei 4 Patienten durchgeführt. Dieser geringe Einsatz der Strahlentherapie bei der Behandlung von Angiosarkomen in unserer Serie lässt sich wahrscheinlich durch die große Anzahl von Fällen von Angiosarkomen nach Bestrahlung in unserer Serie erklären. Mit anderen Worten, sie wurde wahrscheinlich durch ein gewisses Maß an Widerstand gegen die Bestrahlung in diesen Fällen beeinflusst. Der Einsatz der Strahlentherapie bei Angiosarkomen nach Bestrahlung wird in der Literatur jedoch befürwortet und manchmal sogar als Monotherapie ohne Operation durchgeführt.23,24 Bei der Hälfte der Patienten wurde eine Chemotherapie durchgeführt. Wie bereits erwähnt, hatte sie lediglich eine palliative Funktion und war in allen Fällen mit einem schlechten Ergebnis verbunden. Obwohl die Paclitaxel-Monotherapie in unserer Serie nicht das am häufigsten eingesetzte Chemotherapieschema war (da die Fälle vor einiger Zeit diagnostiziert wurden), wird dieses Schema heute in den meisten Fällen als Erstlinienoption eingesetzt. Trotz der anfänglichen Erwartungen werden angiogene Medikamente (Sunitinib, Sorafenib, Bevacizumab, Thalidomid) aufgrund ihrer enttäuschenden Ergebnisse in dieser Situation nicht eingesetzt.
Trotz der geringen Fallzahl in unserer Serie kutaner Angiosarkome, die uns an der Durchführung statistischer Analysen hinderte, stellten wir fest, dass sowohl eine größere Tumorgröße als auch ein höheres Alter mit einer schlechteren Prognose verbunden waren. Ein weniger offensichtlicher Zusammenhang wurde auch zwischen einer schlechten Prognose und den folgenden histologischen Merkmalen beobachtet: Vorhandensein von Nekrosen, Vorherrschen von epitheloiden Zellen, Invasion tieferer Schichten und eine größere Anzahl von Mitosen.
Ethische OffenlegungenSchutz von Mensch und Tier
Die Autoren erklären, dass für diese Studie keine Versuche an Menschen oder Tieren durchgeführt wurden.
Vertraulichkeit der Daten
Die Autoren erklären, dass sie das Protokoll ihres Krankenhauses zur Veröffentlichung von Patientendaten befolgt haben.
Recht auf Privatsphäre und informierte Zustimmung
Die Autoren erklären, dass in diesem Artikel keine privaten Patientendaten erscheinen.
Interessenkonflikte
Die Autoren erklären, dass sie keine Interessenkonflikte haben.