Angiossarcoma cutâneo tem um dos piores prognósticos entre todos os tumores cutâneos. É muito agressivo e tem altas taxas de recidiva local. A taxa de sobrevida em 5 anos está entre 12% e 34% de acordo com a maioria dos estudos,1,2 mas pode chegar a 62%.3 Ao contrário de outros sarcomas, o grau de diferenciação não tem sido associado ao prognóstico do angiossarcoma cutâneo.4
A forma clássica do angiossarcoma cutâneo é uma lesão edematosa mal definida, com uma apresentação clínica largamente indolente em suas fases iniciais. Ocorre na face ou no couro cabeludo de pacientes idosos (angiossarcoma de Wilson-Jones) e representa aproximadamente 50% de todos os angiossarcomas cutâneos primários.5-8 Duas outras formas típicas de angiossarcoma são a síndrome de Stewart-Treves, que se desenvolve em áreas de linfedema de longa duração e é particularmente comum em mulheres que foram submetidas a mastectomia radical,9-11 e o angiossarcoma pós-tradiação, que se desenvolve em áreas de pele irradiada, particularmente na região peitoral de mulheres com histórico de câncer de mama tratado com radioterapia.12-A aparência histopatológica do angiossarcoma cutâneo varia de formas relativamente diferenciadas com espaços vasculares reconhecidamente cobertos por células endoteliais proeminentes com alguma atipia e um padrão infiltrativo dissecando os feixes de colágeno até formas mais sólidas altamente indiferenciadas compostas de células fusiformes ou epitélioides com atipias e pleomorfismos consideravelmente maiores e um número maior de mitoses. Os espaços vasculares são raros e os tumores podem às vezes imitar o carcinoma.
O tratamento principal para o angiossarcoma cutâneo – e o único potencialmente curativo se forem alcançadas margens livres de doença – é a excisão cirúrgica com amplas margens seguida de radioterapia local e até mesmo, na opinião de alguns autores, a radiação dos linfonodos regionais.6 Na maioria dos casos, entretanto, não é fácil alcançar margens livres de doença devido à extensa disseminação subclínica. Além disso, estes tumores são frequentemente multifocais. A quimioterapia tem um papel meramente paliativo no manejo do angiossarcoma cutâneo.
Embora o angiossarcoma cutâneo seja raro (representando menos de 1% de todos os sarcomas), a maioria dos casos de angiossarcoma tem origem na pele. Devido à sua baixa prevalência, os angiossarcomas cutâneos estão incluídos em séries de angiossarcoma visceral ou ósseo, que têm um prognóstico ainda mais sombrio.1 Assim, existem poucas séries grandes de angiossarcoma cutâneo na literatura devido à escassez de casos uniformes a longo prazo.2,4,5,7,16 O manejo do angiossarcoma é, além disso, freqüentemente desanimador, sobretudo nos casos de doença avançada, que têm um prognóstico muito ruim apesar do uso de tratamento agressivo desde o início. Motivados assim pelas dificuldades associadas ao manejo do angiossarcoma cutâneo e pela pouca literatura disponível, investigamos todos os casos de angiossarcoma cutâneo tratados no Instituto Valenciano de Oncologia (IVO), em Valência, Espanha, com o objetivo de identificar fatores clínicos, histológicos e relacionados ao tratamento possivelmente associados ao prognóstico. Para isso, revisamos prontuários médicos e achados clínicos em busca de dados exploratórios que pudessem servir como guia para diagnóstico precoce, já que pacientes com doença precoce e pequenos tumores têm uma probabilidade de sobrevida consideravelmente melhorada.
Material e Métodos
Realizamos um estudo observacional retrospectivo de todos os casos de angiossarcoma cutâneo tratados na IVO entre janeiro de 2000 e dezembro de 2015. Toda a informação compilada foi extraída dos prontuários médicos dos pacientes, do arquivo de biópsias do departamento de patologia e do arquivo fotográfico do nosso departamento. Dos 20 casos inicialmente identificados, 4 tiveram que ser excluídos: 1 porque não houve seguimento, outro porque não havia material suficiente para determinar se o tumor era um hemangioendotelioma ou um angiossarcoma, e 2 porque os tumores não eram angiossarcomas primários. Estes 2 tumores tinham sido inicialmente rotulados como angiossarcoma cutâneo porque todas as lâminas de histologia mostravam envolvimento cutâneo da mama. No entanto, ao revisar os bloqueios, descobrimos que o envolvimento cutâneo era secundário em ambos os casos, e que o tumor primário estava localizado no parênquima mamário de onde se estendia até a pele sobrejacente.
Os critérios de inclusão no estudo foram achados clínicos sugestivos de angiossarcoma cutâneo e confirmação histológica do diagnóstico com coloração de hematoxilina-eosina de amostras de biópsia, suportados por estudos imunohistoquímicos, que na maioria dos casos envolveram a coloração de CD31, CD34, D240 e Ki-67.
Seis casos de angiossarcoma cutâneo foram incluídos no estudo. Corresponderam a 11 mulheres e 5 homens entre 35 e 83 anos (média, 67 anos; mediana, 71 anos). Dez dos casos foram angiossarcoma pós-tradiação (10 casos), 5 foram angiossarcoma de Wilson-Jones e apenas 1 foi angiossarcoma associado à linfedema. O local mais comum foi o tronco (10 casos), seguido pela cabeça e pescoço (5 casos). As extremidades superiores estavam envolvidas em apenas 1 caso. O menor tamanho do tumor era de 1cm e o maior de 50cm (média, 10cm; mediana, 6,5cm).
Onze das pacientes tinham história de câncer (câncer de mama em 10 casos e seminoma em 1). Com exceção de 1 caso de carcinoma lobular invasivo, todos os cânceres de mama eram carcinomas ductais invasivos.
O tempo médio entre a radioterapia e o desenvolvimento de angiossarcoma nos 10 casos de angiossarcoma pós-tradiação foi de 8,2 anos. Apenas 1 dos casos apareceu dentro de 5 anos após a radioterapia; os demais apareceram pelo menos 5 anos depois.
Quatorze casos foram tratados cirurgicamente e a radioterapia adjuvante foi utilizada em 4 deles. Oito pacientes receberam quimioterapia e este foi o primeiro e único tratamento em 2 pacientes.
Doxorubicina e taxol foram utilizados em 4 casos cada, ifosfamida em 3 casos e paclitaxel e dacarbazina em 1 caso cada. A resposta à quimioterapia foi pobre e, embora quase todos os pacientes tenham apresentado resposta parcial, a doença progrediu em todos os casos e os pacientes morreram durante o acompanhamento (8/8).
Cinco pacientes apresentaram metástases à distância, o que envolveu múltiplos locais na maioria dos casos. Os locais mais comuns foram o pulmão e o fígado.
Dez dos 16 pacientes morreram de angiossarcoma durante o acompanhamento. Os outros 6 pacientes estão atualmente livres de doença. A duração média do seguimento foi de 42,5 meses (mediana, 26 meses; variação, 7-188 meses).
Histologicamente, 8 casos tiveram um padrão de crescimento sólido, 4 tiveram um padrão vasoformativo, e 4 tiveram um padrão misto. O tipo celular predominante foi o epitélioide em 14 casos e o fuso em apenas 2. A necrose foi observada em 6 tumores e o padrão infiltrativo foi subcutâneo na maioria dos casos (n=10). Quatro casos foram confinados à derme e apenas 2 afetaram os planos musculares. As margens cirúrgicas não puderam ser avaliadas em 3 casos. Dos casos restantes, 8 tinham margens negativas e 5 tinham margens positivas. A reação linfocítica foi leve ou moderada em 10 casos, intensa em 2, e inexistente em 4. Nos 14 casos com reação linfocítica, o infiltrado foi peritumoral em 2 casos, intratumoral em 8, e misto em 2. Houve média de 15 mitoses por 10 campos (variação, 0-37 mitoses).
Os resultados clínicos e patológicos mais relevantes estão resumidos na Tabela 1. Os resultados da comparação entre sobreviventes e não sobreviventes estão resumidos na Tabela 2.
Selecção de Resultados Clínicos e Patológicos para os 16 Angiossarcomas Cutâneos.a
| Patiente | Age, y | Sexo | Tipo | Localização | Tamanho, cm | Tempo Desde Rx, mo | Dose, Gy | Tumor anterior | Breast Cancer Type | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | F | PR | Peito esquerdo | 18 | 60 | 46 | Peito | IDC | ||
| 2 | 71 | ST | Braço esquerdo | 12 | – | – | Mama | IDC | |||
| 3 | 51 | F | PR | Região submamária direita | 1 | 57 | 50 | >Pernas | IDC | ||
| 4 | > 76 | F | WJ | Cabeça e pescoço | 3 | – | – | Não | . | ||
| >5 | 77 | F | PR | Peito direito | 1 | 94 | 46 | Peito | IDC | ||
| 6 | 71 | F | PR | Peito esquerdo | 50 | 171 | 48 | Peito | IDC | ||
| 7 | 48 | M | PR | Muro abdominal | 2 | 96 | 26 | Seminoma | – | ||
| 8 | 55 | F | PR | Peito esquerdo | 10 | 88 | 46 | Peito | ILC | ||
| 9 | 69 | F | PR | Peito esquerdo | 8 | 143 | 46 | Peito | IDC | ||
| 10 | 76 | M | WJ | Bochecha direita | 6 | – | – | – | Não | – | |
| >11 | 35 | F | PR | Seios rectos | 12 | 66 | 50 | Breast | IDC | ||
| 12 | 57 | F | PR | Peito direito | 8 | 108 | 50 | Peito | IDC | ||
| 13 | 68 | M | WJ | Cabeça e pescoço | 2 | – | – | – | – | ||
| 14 | 80 | M | WJ | Cabeça e pescoço | 15 | – | – | Não | – | ||
| >15 | 79 | M | WJ | Cabeça e pescoço | 2 | – | – | Não | – | ||
| >16 | 83 | F | PR | Samo esquerdo | 3 | 110 | 50 | Breast | IDC | ||
| X=67.1 | 11W, 5M | 10 RI, 5 WJ, 1 ST | 9 peito, 5 cabeça e pescoço, 1 abdómen, 1 membro superior | X=10 | X=100.3 | X=45.8 | 10 peito, 1 seminoma |
10 cancros do peito: 9 IDC, 1 ILC |
| Patiente | Cirurgia | Margin, cm | Tratamento como Tratamento | Morte | PadrãoHP | Tipo de célula | Necrose | DoI | Mitoses/mm2 | Survival, mo | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Sim | 0.2 | 13 | Sim | 1 | E | Não | 3 | 7 | 24 | ||
| 2 | Não | ns | 3 | Sim | 2 | > E | Não | 3 | 37 | 8 | ||
| 3 | Sim | Sim | 3.5 | >1 | Não | 1 | E | Não | 3 | 2 | 29 | |
| 4 | Sim | 2 | 123 | Sim | 2 | E | Não | 3 | 28 | 26 | ||
| 5 | Sim | ns | 1 | Sim | 2 | E | Não | 2 | 14 | 8 | ||
| 6 | Não | ns | 3 | Sim | 1 | E | Não | 2 | 0 | 19 | ||
| 7 | Yes | >ns | 1 | Não | 3 | E | >Não | 3 | 6 | 187 | ||
| 8 | Sim | ns | 13 | Sim | 2 | E | Sim | 4 | 6 | 28 | ||
| 9 | Sim | ns | 13 | Sim | 3 | E | Sim | 3 | 22 | 24 | ||
| 10 | Sim | ns | 12 | Sim | 2 | E | Sim | 3 | 16 | 7 | ||
| 11 | Sim | >0.5 | 1 | Não | 2 | >SC | >Não | 3 | 18 | 76 | ||
| >12 | Sim | 0 | 13 | Sim | 3 | E | Sim | 3 | 36 | 26 | ||
| 13 | Sim | 2 | 12 | Não | 3 | >E | Não | 3 | 5 | 95 | ||
| 14 | Sim | 2 | 123 | Sim | 2 | E | Sim | 4 | 17 | 24 | ||
| >15 | Sim | 2 | 1 | Não | 2 | E | Sim | 2 | 6 | 53 | ||
| 16 | Sim | 3 | 1 | Não | 1 | SC | Não | 2 | 23 | 51 | ||
| 14 sim, 2 não | 1.68 | 14: Qx 4: Rt 8: Qt |
10 sim 6 não |
4 vasof 8 sólido 4 misturado |
14 E, 2 SC | 6 Sim 10 Não |
4 derme 10 hipoderme 2 músculo |
X=15 | X=42.8 |
Os pacientes que morreram são mostrados em negrito.
Tratamento por E, tratamento de angiossarcoma (1, cirurgia; 2, radioterapia; 3, quimioterapia); DoI, profundidade de invasão (2, derme; 3, hipoderme; 4, músculo); E, epitélioide; HP, histopatológico (1, vasoformativa; 2, sólido; 3, misto); IDC, carcinoma ductal invasivo; ILC, carcinoma lobular invasivo; M, homem; ns, não especificado; PR, angiossarcoma pós-tradiação; Rx, radiação; SC, fuso celular; ST, Stewart-Treves angiossarcoma; W, mulher; WJ, Wilson-Jones angiossarcoma: X, média.
Comparação de Variáveis entre Sobreviventes e Pacientes que Morreram de Angiossarcoma Cutâneo.
| Variável | Vivos (n=6) | Mortos (n=10) | |
|---|---|---|---|
| Idade, média, y | 61 | 71 | |
| Mulheres | 3 | 8 | |
| > Homens | 3 | 2 | |
| Postradiação | 4 | 6 | |
| Idiopático | 2 | 3 | |
| Linfedema…associado | 0 | 1 | |
| Tronco | 4 | 6 | |
| Cabeça e pescoço | 2 | 3 | |
| Membros superiores | 0 | 1 | |
| Tamanho, cm | 3.6 | 13.1 | |
| Tempo desde a radioterapia, mo | 82.25 | 110.6 | |
| Dose de radiação, Gy | 44 | 47 | |
| > Peito | 3 | 7 | |
| Seminoma | 1 | ||
| Cirurgia | 6 | 8 | |
| Radioterapia | 1 | 3 | |
| Quimioterapia | 0 | 8 | |
| Vasoformativo | 2 | 2 | |
| Sólido | 2 | 6 | |
| Misturado | 2 | 2 | |
| Necrose | 1 | 5 | |
| Derme | 2 | 2 | |
| Subcutâneo | 4 | 6 | |
| Músculo | 0 | 2 | |
| Mitoses | 10 | 18.3 | |
| Sobrevivência, mo | 81.8 | 19.4 | |
Discussão
Angiossarcoma cutâneo é um tumor muito raro, evidenciado pelo facto de só termos sido capazes de recolher dados sobre 16 tumores diagnosticados durante um período de 14 anos num hospital oncológico. Em geral, o angiossarcoma cutâneo é um pouco mais comum em homens idosos. Isto porque a forma mais comum de angiossarcoma cutâneo na população geral é o angiossarcoma primário de cabeça e pescoço (também conhecido como idiopático ou angiossarcoma de Wilson-Jones)(Fig. 1), que tende a afetar os homens idosos.5,7,17 O angiossarcoma de postradiação (Fig. 2) é agora a segunda forma mais comum de angiossarcoma cutâneo devido ao uso crescente de radioterapia em oposição à mastectomia radical para tratar o câncer de mama.2,13,18 Esta alteração também levou a uma redução na frequência do angiossarcoma associado à linfedema, que é atualmente a forma menos comum deste tumor. A prevalência das diferentes formas de angiossarcoma cutâneo em nossa série não coincide com os relatos da literatura, pois nosso hospital trata um grande número de mulheres com câncer de mama, explicando porque a forma mais comum de angiossarcoma cutâneo em nosso hospital foi o angiossarcoma pós-tradiação. Esta predominância de casos de câncer de mama também explica o porquê de 11 das 16 pacientes serem mulheres. Houve apenas 1 caso de angiossarcoma associado à linfedema (Fig. 3), o que coincide com as taxas de prevalência relatadas em outros lugares. A paciente teve linfedema crônico do braço esquerdo secundário à dissecção dos linfonodos axilares realizada como parte do tratamento do câncer de mama 22 anos antes.
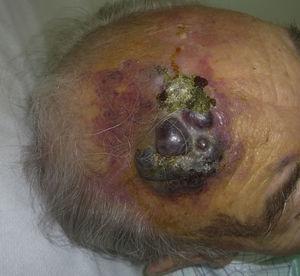
Chapa vermelha-violácea com áreas nodulares na testa de um homem idoso.
>
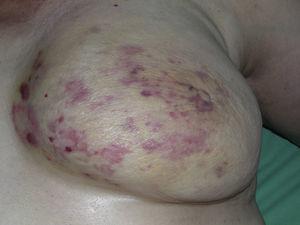
Máculas múltiplas e manchas eritematosas com várias pápulas avermelhadas em pele amarelada sobre um seio irradiado devido ao cancro.
>
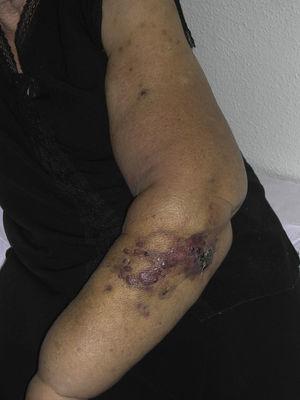
Pápulas vermelhas-violáceas e nódulos com áreas semelhantes a hematomas num braço com linfedema secundário à cirurgia do cancro da mama.
>
Com excepção de 1 mulher que teve angiossarcoma idiopático ou Wilson-Jones, o resto das mulheres da nossa série (n=10) tinham uma história de cancro da mama. Nove destes cancros eram carcinomas ductais invasivos. Como 80% dos cânceres de mama na população geral são carcinomas ductais invasivos e apenas 10% são carcinomas lobulares, a alta prevalência de carcinoma ductal invasivo em nosso estudo é provavelmente um reflexo da alta prevalência deste câncer na população geral, e não de uma associação particular entre angiossarcoma cutâneo pós-tradiação e carcinoma ductal invasivo, como nossos resultados parecem sugerir. Em outras palavras, as proporções observadas em nossa série estão em consonância com as taxas descritas para diferentes cancros da mama na população em geral. Houve 2 casos de angiossarcoma mamário inicialmente incluídos em nosso estudo que merecem ser mencionados. Foi relatado que o angiossarcoma mamário tende a ser cutâneo quando induzido por radiação e parenquimatoso quando não é.19 Em nossa série, dos 18 casos de angiossarcoma cutâneo inicialmente identificados, houve 2 casos de angiossarcoma mamário não induzidos por radioterapia. Ao rever esses casos, descobrimos que a localização primária do tumor era o parênquima mamário e não a pele. Os tumores foram, portanto, excluídos do estudo por serem secundários e não primários. Os casos restantes foram todos angiossarcomas mamários primários induzidos por radiação, de acordo com relatos da literatura.
O local mais comum para angiossarcoma em nossa série foi a mama (10 casos) e não a face ou couro cabeludo, como seria de se esperar. O único caso que envolveu as extremidades foi um angiossarcoma associado a uma linfedema. Novamente, o fato do local mais comum do tumor ser a mama seria explicado pela predominância de tumores induzidos pela radiação em nossa série.
Apesar de o período de latência entre a exposição à radioterapia e o desenvolvimento do angiossarcoma ser altamente variável de acordo com relatos na literatura (3-50 anos), tende a ser mais longo (em média, 25 anos) quando a doença tratada pela radiação é benigna. Os períodos de latência relatados para doenças malignas são mais curtos (cerca de 10-15 anos), exceto no caso do angiossarcoma da mama, para o qual foi descrita uma média de aproximadamente 5 anos.12 A razão para este período mais curto no caso do angiossarcoma de mama não é clara, embora várias teorias tenham sido propostas, incluindo o grande volume de pele irradiada, a presença de linfedema associado, fatores possivelmente intrínsecos à mama e um possível efeito sinérgico com a quimioterapia.13,14 Em nossos 10 casos pós-tradiação, o angiossarcoma foi diagnosticado após uma média de 8,2 anos, e pelo menos 5 anos tinham passado em 90% dos casos. Nenhuma associação entre o período de latência e o prognóstico foi relatada para o angiossarcoma. Em nossa série, o tempo médio desde a radioterapia até o desenvolvimento do angiossarcoma foi um pouco maior nos pacientes que morreram (110,6 meses) do que naqueles que sobreviveram (82,25 meses). No único caso da síndrome de Stewart-Treves, o paciente tinha sido submetido a dissecção dos gânglios linfáticos 22 anos antes. Os períodos de latência relatados para o angiossarcoma associado à linfedema são altamente variáveis, com uma variação de 1 a 30 anos e uma média de 10. A síndrome de Stewart-Treves representa 90% de todos os casos de angiossarcoma associado a linfedema.9,11 No entanto, o angiossarcoma também pode surgir em outras formas de linfedema, como linfedema congênito, linfedema filarial e linfedema secundário à dissecção dos linfonodos em outras partes do corpo.10
O tamanho do tumor é atualmente o marcador prognóstico mais amplamente aceito para o angiossarcoma cutâneo, e tem sido frequentemente relatado que angiossarcomas medindo 5cm ou mais têm pior prognóstico do que tumores menores.5,17 Em nossa série, observamos diferenças no tamanho médio do tumor entre sobreviventes e não sobreviventes. Aqueles que sobreviveram tiveram um tamanho médio do tumor de 3,6cm, que foi mais de 3 vezes menor do que o tamanho médio (13,1cm) no grupo de pacientes que morreram. Nossos resultados, portanto, sustentam a crença de que tumores maiores estão associados a piores resultados no angiossarcoma e destacam a importância de diagnosticar o tumor quando este começa como uma lesão tipo contusão, embora isto seja particularmente difícil nos angiossarcomas do couro cabeludo e ainda mais quando o paciente ainda tem cabelos (Fig. 4). O angiossarcoma da mama por via postradiátrica é mais fácil de diagnosticar, já que qualquer lesão persistente nesta área com aspecto vascular ou tipo hematoma deve ser biopsiada. Uma pista diagnóstica potencialmente útil é um halo amarelado (correspondente à hemossiderina) ao redor da lesão suspeita (Fig. 5), pois nunca observamos este sinal em proliferações vasculares benignas em pele irradiada. Postradiation angiosarcoma and atypical vascular proliferations on irradiated skin must be distinguished histologically but clinical features can help. As proliferações vasculares atípicas tendem a ser muito menores do que os angiossarcomas e geralmente têm um período de latência mais curto.20 Ao contrário dos angiossarcomas, estas proliferações estão confinadas à derme superficial e média e não invadem o tecido subcutâneo. Além disso, a histologia não mostra a atipia nuclear característica observada no angiossarcoma, nem as múltiplas camadas de células endoteliais ou figuras mitóticas. No entanto, ocasionalmente pode ser difícil distinguir entre as 2 entidades e há relatos de 2 lesões coexistentes na mesma mama irradiada. Houve mesmo casos de proliferações vasculares atípicas induzidas por radiação que progrediram para o angiossarcoma.21 Em casos muito complicados, pode ser útil buscar a superexpressão do gene MYC se for realizado um estudo imunohistoquímico, ou a amplificação se for usada a hibridação fluorescente in situ. A amplificação de MYC é bastante comum em angiossarcomas secundários, mas não parece ocorrer em proliferações vasculares atípicas em pele irradiada.22
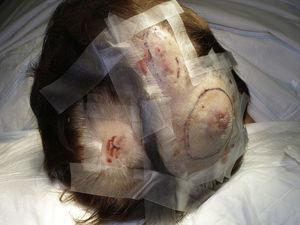
Nódulos eritematosos multifocais no couro cabeludo.
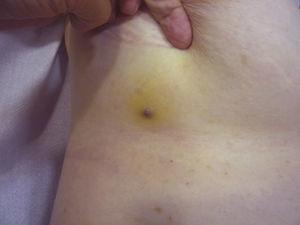
Pápula vermelha-violácea medindo menos de 1cm cercada por uma área amarelada da pele.
O outro marcador prognóstico mais amplamente aceito no angiossarcoma é a idade. Em nossa série, os pacientes que sobreviveram eram em média mais jovens do que aqueles que morreram (62 contra 71 anos). Apesar disso, 10 dos 16 pacientes de nossa série haviam morrido ao final do estudo, corroborando o mau prognóstico geralmente associado ao angiossarcoma.
A exemplo de outros sarcomas, o Comitê Conjunto Americano de Câncer não considera o grau histológico como um marcador prognóstico relevante no angiossarcoma. Vários autores, entretanto, têm sugerido que certas características histopatológicas podem ter influência no prognóstico. Alguns estudos recentes, por exemplo, afirmam que um padrão sólido predominante pode ser um marcador prognóstico relativamente bom no angiossarcoma da cabeça e pescoço.3,16 Este não foi o caso na nossa série, pois 6 dos 10 pacientes que morreram tinham este padrão, comparado com apenas 2 dos 6 pacientes que ainda estavam vivos no final do estudo (Figs. 6 e 7).
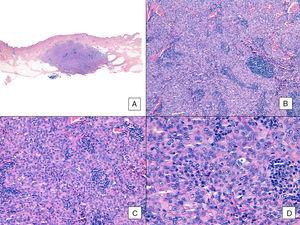
Angiossarcoma com padrão sólido predominante. A, A imagem panorâmica mostra invasão da derme reticular média e profunda e da hipoderme (hematoxilina-eosina, ampliação original ×10). B, Tumor densamente celular que destruiu estruturas pré-existentes e é acompanhado por infiltrados linfóides nodulares (hematoxilina-eosina, ampliação original ×100). C e D, Vista detalhada mostrando uma predominância de células epitelioides acompanhadas por um infiltrado linfocitário (hematoxilina-eosina ×200 e ×400, respectivamente).
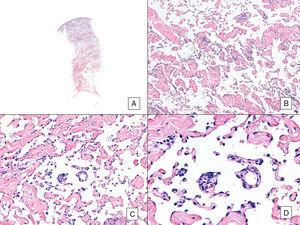
Angiossarcoma com padrão vasoformativo predominante. A, Imagem panorâmica mostrando infiltração da derme até a hipoderme (hematoxilina-eosina, ampliação original ×10). B, Espaços vasculares neoplásicos com dissecção pronunciada de colágeno (hematoxilina-eosina, ampliação original ×100). C e D, vasos não-neoplásicos dissecados por células endoteliais neoplásicas, que permanecem “flutuando” na derme (signo promontório) (hematoxilina-eosina, ampliação original ×200 e ×400, respectivamente).
Outros fatores histológicos potencialmente de alto risco propostos na literatura são a presença de necrose, uma morfologia epitélioide e uma maior profundidade de invasão.5,7 Os nossos resultados apoiam estes potenciais marcadores, uma vez que as 3 variáveis foram mais comuns no grupo dos não-sobreviventes. A necrose foi observada em 5 dos 10 pacientes que morreram e em apenas 1 dos 6 pacientes que sobreviveram. Da mesma forma, os únicos 2 pacientes com padrão predominante de células fusiformes ainda estavam vivos no final do estudo. Nenhum dos pacientes que faleceram, pelo contrário, tinha este padrão. Finalmente, os únicos 2 pacientes com envolvimento dos planos musculares morreram. O número médio de mitoses também foi maior no grupo de não-sobreviventes (18,3 vs 10).
O tratamento de escolha do angiossarcoma cutâneo é a excisão cirúrgica com amplas margens seguida de radioterapia.6 A frequente natureza multifocal do angiossarcoma e sua propensão para o desenvolvimento de propagação subclínica muitas vezes complica a obtenção de margens claras. Além disso, não há consenso universal sobre o que constitui uma margem adequada, e a maioria dos estudos fornece informações imprecisas, como a excisão com “amplas margens”. Na nossa série, a cirurgia foi o tratamento de escolha em 14 dos 16 pacientes. Margens de 3cm foram utilizadas onde tecnicamente viável e, nos outros casos, tentou-se a desobstrução com margens de 2 cm. Os 14 pacientes foram submetidos a cirurgia radical. Os outros 2 não foram considerados candidatos à cirurgia e receberam quimioterapia paliativa. Eles morreram durante o acompanhamento. A radioterapia adjuvante ao leito cirúrgico foi utilizada em apenas 4 pacientes. Este baixo uso de radioterapia no manejo do angiossarcoma em nossa série provavelmente pode ser explicado pelo grande número de casos de angiossarcomas pós-tradiação em nossa série. Em outras palavras, provavelmente foi influenciado por um certo nível de resistência ao uso de radiação nestes casos. O uso de radioterapia no angiossarcoma pós-tradiação, no entanto, encontra apoio na literatura, sendo até, por vezes, administrado como monoterapia, sem cirurgia.23,24 A quimioterapia foi utilizada em metade dos pacientes. Como já mencionado, ela teve um papel meramente paliativo e foi associada a um mau resultado em todos os casos. Embora a monoterapia com paclitaxel não fosse o regime de quimioterapia mais utilizado na nossa série (pois os casos foram diagnosticados há algum tempo), este regime é agora utilizado como uma opção de primeira linha na maioria dos casos. Apesar das expectativas iniciais geradas, drogas angiogênicas (sunitinib, sorafenib, bevacizumab, talidomida) não são utilizadas neste ambiente devido aos seus resultados decepcionantes.
Embora o pequeno número de casos em nossa série de angiossarcoma cutâneo, que nos impediu de realizar análises estatísticas, observamos que tanto o maior tamanho do tumor quanto a idade mais avançada estavam associados a pior prognóstico. Uma associação menos evidente também foi observada entre o mau prognóstico e as seguintes características histológicas: presença de necrose, predominância de células epiteliais, invasão de camadas mais profundas e maior número de mitoses.
Divulgação ÉticaProteção de seres humanos e animais
Os autores declaram que não foram realizados testes em seres humanos ou animais para os fins deste estudo.
Confidencialidade dos dados
Os autores declaram ter seguido o protocolo do seu hospital sobre a publicação de dados relativos a pacientes.
Direito à privacidade e consentimento informado
Os autores declaram que não aparecem dados de pacientes particulares neste artigo.
Conflitos de interesse
Os autores declaram não ter conflitos de interesse.