Kutánní angiosarkom má jednu z nejhorších prognóz mezi všemi kožními nádory. Je velmi agresivní a má vysokou míru lokálních recidiv. Pětileté přežití se podle většiny studií pohybuje mezi 12 % a 34 %,1,2 ale může dosahovat až 62 %.3 Na rozdíl od jiných sarkomů nebyl u kožního angiosarkomu stupeň diferenciace spojen s prognózou.4
Klasická forma kožního angiosarkomu je špatně definovaná edematózní léze podobná modřině s převážně indolentním klinickým obrazem v časných fázích. Vyskytuje se na obličeji nebo pokožce hlavy u starších pacientů (Wilsonův-Jonesův angiosarkom) a tvoří přibližně 50 % všech primárních kožních angiosarkomů.5-8 Dalšími dvěma typickými formami angiosarkomu jsou Stewartův-Trevesův syndrom, který vzniká v oblastech dlouhodobého lymfedému a je zvláště častý u žen, které podstoupily radikální mastektomii,9-11 a postradiační angiosarkom, který vzniká v oblastech ozářené kůže, zejména v pektorální oblasti u žen, které v minulosti prodělaly karcinom prsu léčený radioterapií.12-15
Histopatologický vzhled kožního angiosarkomu kolísá od relativně diferencovaných forem s rozpoznatelnými cévními prostory pokrytými prominujícími endoteliálními buňkami s určitou atypií a infiltrativním vzorem disekujícím kolagenní svazky až po solidnější vysoce nediferencované formy složené z vřetenitých nebo epiteloidních buněk s podstatně větší atypií a pleomorfismem a vyšším počtem mitóz. Cévní prostory jsou vzácné a nádory mohou někdy napodobovat karcinom.
Základní léčbou kožního angiosarkomu – a jedinou, která je potenciálně kurativní, pokud je dosaženo okrajů bez onemocnění – je chirurgická excize s širokými okraji následovaná lokální radioterapií a podle některých autorů dokonce ozářením regionálních lymfatických uzlin.6 Ve většině případů však není snadné dosáhnout okrajů bez onemocnění kvůli rozsáhlému subklinickému šíření. Kromě toho jsou tyto nádory často multifokální. Chemoterapie má v léčbě kožního angiosarkomu čistě paliativní úlohu.
Ačkoli je kožní angiosarkom vzácný (tvoří méně než 1 % všech sarkomů), většina případů angiosarkomu vzniká v kůži. Vzhledem k nízkému výskytu jsou kožní angiosarkomy zařazovány do sérií viscerálních nebo kostních angiosarkomů, které mají ještě neutěšenější prognózu.1 V literatuře tak existuje jen málo velkých sérií kožních angiosarkomů z důvodu nedostatku dlouhodobých jednotných případů.2,4,5,7,16 Management angiosarkomů je navíc často skličující, především v případech pokročilého onemocnění, které mají velmi špatnou prognózu i přes použití agresivní léčby od počátku. Motivováni obtížemi spojenými s léčbou kožního angiosarkomu a malým množstvím dostupné literatury jsme proto zkoumali všechny případy kožního angiosarkomu léčené v Instituto Valenciano de Oncología (IVO) ve španělské Valencii s cílem identifikovat klinické, histologické a s léčbou související faktory, které by mohly souviset s prognózou. Za tímto účelem jsme prošli lékařské záznamy a klinické nálezy a hledali jsme explorativní údaje, které by mohly sloužit jako vodítko pro včasnou diagnózu, protože pacienti s časným onemocněním a malými nádory mají výrazně vyšší pravděpodobnost přežití.
Materiál a metody
Provedli jsme retrospektivní observační studii všech případů kožního angiosarkomu léčených na IVO v období od ledna 2000 do prosince 2015. Veškeré shromážděné informace byly získány ze zdravotnické dokumentace pacientů, z archivu biopsií patologického oddělení a z fotografického archivu našeho oddělení. Z původně identifikovaných 20 případů musely být 4 vyřazeny: 1 z nich nebyl sledován, další proto, že nebyl k dispozici dostatečný materiál k určení, zda se jedná o hemangioendoteliom nebo angiosarkom, a 2 proto, že se nejednalo o primární angiosarkom. Tyto 2 nádory byly původně označeny jako kožní angiosarkom, protože všechny histologické preparáty vykazovaly kožní postižení prsu. Při revizi bloků jsme však zjistili, že kožní postižení bylo v obou případech sekundární a že primární nádor se nacházel v mamárním parenchymu, odkud zasahoval až do nadloží.
Kritériem pro zařazení do studie byly klinické nálezy svědčící pro kožní angiosarkom a histologické potvrzení diagnózy pomocí barvení vzorků biopsie hematoxylinem-eozinem, podpořené imunohistochemickým vyšetřením, které ve většině případů zahrnovalo barvení CD31, CD34, D240 a Ki-67.
U každého pacienta byly studovány následující proměnné: věk, pohlaví, umístění a velikost nádoru, typ angiosarkomu (primární, postradiační, asociovaný s lymfedémem), léčba (operace, radioterapie, chemoterapie), recidiva, metastázy, přežití a úmrtí. U postradiačních a s lymfedémem asociovaných nádorů jsme zaznamenávali také typ předchozího nádoru a počet let od radioterapie nebo lymfedému. Analyzovanými histologickými proměnnými byly stav okrajů, histopatologický vzorec (vazoformní, solidní nebo smíšený), převažující typ buněk (epiteloidní nebo vřetenobuněčný), přítomnost nekrózy (ano, ne), úroveň invaze (epidermis, dermis, hypodermis, sval, kost), lymfocytární reakce, infiltrativní vzorec a počet mitóz na 10 polí.
Výsledky
Do studie bylo zahrnuto šestnáct případů kožního angiosarkomu. Odpovídaly 11 ženám a 5 mužům ve věku od 35 do 83 let (průměr 67 let; medián 71 let). V deseti případech se jednalo o postradiační angiosarkom (10 případů), v pěti případech o Wilson-Jonesův angiosarkom a pouze v jednom případě o angiosarkom spojený s lymfedémem. Nejčastější lokalizací byl trup (10 případů), následovaný hlavou a krkem (5 případů). Horní končetiny byly postiženy pouze v 1 případě. Nejmenší velikost nádoru byla 1 cm a největší 50 cm (průměr 10 cm; medián 6,5 cm).
Jedenáct pacientů mělo v anamnéze nádorové onemocnění (karcinom prsu v 10 případech a seminom v 1 případě). S výjimkou 1 případu invazivního lobulárního karcinomu byly všechny karcinomy prsu invazivní duktální karcinomy.
Průměrná doba mezi radioterapií a vznikem angiosarkomu v 10 případech postradiačního angiosarkomu byla 8,2 roku. Pouze 1 z případů se objevil do 5 let od radioterapie, ostatní se objevily nejméně o 5 let později.
Čtrnáct případů bylo léčeno chirurgicky a u 4 z nich byla použita adjuvantní radioterapie. Osm pacientů podstoupilo chemoterapii a u 2 pacientů to byla první a jediná léčba.
Doxorubicin a taxol byly použity každý ve 4 případech, ifosfamid byl použit ve 3 případech a paklitaxel a dakarbazin byly použity každý v 1 případě. Odpověď na chemoterapii byla špatná, a přestože téměř všichni pacienti vykazovali částečnou odpověď, onemocnění ve všech případech progredovalo a pacienti během sledování zemřeli (8/8).
Pět pacientů mělo vzdálené metastázy, které ve většině případů zahrnovaly více míst. Nejčastějšími místy byly plíce a játra.
Deset z 16 pacientů zemřelo v průběhu sledování na angiosarkom. Dalších 6 pacientů je v současné době bez známek onemocnění. Průměrná doba sledování byla 42,5 měsíce (medián 26 měsíců; rozmezí 7-188 měsíců).
Histologicky mělo 8 případů solidní růstový vzorec, 4 vasoformní vzorec a 4 smíšený vzorec. Převažující buněčný typ byl epiteloidní ve 14 případech a vřetenitý pouze ve 2. Nekróza byla pozorována u 6 nádorů a infiltrativní vzorec byl ve většině případů subkutánní (n=10). Čtyři případy byly omezeny na dermis a pouze 2 postihly svalové roviny. Ve 3 případech nebylo možné chirurgické okraje hodnotit. Ze zbývajících případů mělo 8 negativní okraje a 5 pozitivní okraje. Lymfocytární reakce byla mírná nebo středně silná v 10 případech, intenzivní ve 2 a neexistující ve 4. Ve 14 případech s lymfocytární reakcí byl infiltrát peritumorální ve 2 případech, intratumorální v 8 a smíšený ve 2. Na 10 polí bylo průměrně 15 mitóz (rozmezí 0-37 mitóz).
Nejvýznamnější klinické a patologické výsledky jsou shrnuty v Tabulce 1. Výsledky srovnání přeživších a nepřeživších pacientů jsou shrnuty v tabulce 2.
Výběr klinických a patologických výsledků u 16 kožních angiosarkomů.a
| Pacient | Věk, y | Pohlaví | Typ | Lokalita | Velikost, cm | Čas od Rx, mo | Dávka, Gy | Předchozí nádor | Typ karcinomu prsu | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 78 | F | PR | Levý prs | 18 | 60 | 46 | Prs | IDC | |
| 2 | 71 | F | ST | Levá paže | 12 | – | -. | Prs | IDC | |
| 3 | 51 | F | PR | Pravá submamární oblast | 1 | 57 | 50 | Prs. | IDC | |
| 4 | 76 | F | WJ | Hlava a krk | 3 | – | – | Ne | . | |
| 5 | 77 | F | PR | Pravý prs | 1 | 94 | . | 46 | Prs | IDC |
| 6 | 71 | F | PR | Levý prs | 50 | 171 | 48 | Prs | IDC | |
| 7 | 48 | M | PR | Břišní stěna | 2 | 96 | 26 | Seminom | – | |
| 8 | 55 | F | PR | Levý prs | 10 | 88 | 46 | Prs | ILC | |
| 9 | 69 | F | PR | Levý prs | 8 | 143 | 46 | Prs | IDC | |
| 10 | . 76 | M | WJ | Pravá tvář | 6 | – | – | Ne | -. | |
| 11 | 35 | F | PR | Pravý prs | 12 | 66 | 50 | Prsa | IDC | |
| 12 | 57 | F | PR | Pravý prs | 8 | 108 | 50 | Prs | IDC | |
| 13 | 68 | M | WJ | Hlava a krk | 2 | – | – | Ne | – | |
| 14 | 80 | M | WJ | Hlava a krk | 15 | – | -. | Ne | – | |
| 15 | 79 | M | WJ | Hlava a krk | 2 | – | – | No | – | |
| 16 | 83 | F | PR | Levý prs | 3 | 110 | 50 | Prsa | IDC | |
| X=67.1 | 11W, 5M | 10 RI, 5 WJ, 1 ST | 9 prsou, 5 hlavy a krku, 1 břicha, 1 horní končetiny | X=10 | X=100.3 | X=45.8 | 10 karcinomů prsu, 1 seminom |
10 karcinomů prsu: 9 IDC, 1 ILC |
| Pacient | Chirurgie | Margin, cm | AS léčba | Úmrtí | HP vzor | Typ buňky | Nekróza | DoI | Mitozy/mm2 | Přežití, mo | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Ano | 0.2 | 13 | Ano | 1 | E | Ne | 3 | 7 | 24 | |
| 2 | Ne | ns | 3 | Ano | 2 | E | Ne | 3 | 37 | 8 | |
| 3 | Ano | 3.5 | 1 | Ne | 1 | E | Ne | 3 | 2 | 29 | |
| 4 | Ano | 2 | . | 123 | Ano | 2 | E | Ne | 3 | 28 | 26 |
| 5 | Ano | ns | 1 | Ano | 2 | E | Ne | 2 | 14 | 8 | |
| 6 | Ne | ns | 3 | Ano | 1 | E | Ne | 2 | 0 | 19 | |
| 7 | Ano | ns | 1 | Ne | 3 | E | Ne | 3 | 6 | 187 | |
| 8 | Ano | ns | 13 | Ano | 2 | E | Ano | 4 | 6 | 28 | |
| 9 | Ano | ns | 13 | Ano | 3 | E | Ano | 3 | 22 | 24 | |
| 10 | Ano | ns. ns | 12 | Ano | 2 | E | Ano | 3 | 16 | 7 | |
| 11 | Ano | 0.5 | 1 | Ne | 2 | SC | Ne | 3 | 18 | 76 | |
| 12 | Ano | 0 | 13 | Ano | 3 | E | Ano | 3 | 36 | 26 | |
| 13 | Ano | 2 | 12 | No | 3 | E | Ne | 3 | 5 | 95 | |
| 14 | Ano | 2 | 123 | Ano | 2 | E | Ano | 4 | 17 | 24 | |
| 15 | Ano | 2 | 1 | Ne | 2 | E | Ano | 2 | 6 | 53 | |
| 16 | Ano | 3 | 1 | Ne | 1 | SC | Ne | 2 | 23 | 51 | |
| 14 ano, 2 ne | 1.68 | 14: Qx 4: Rt 8: Qt |
10 ano 6 ne |
4 vasof 8 solid 4 mixed |
14 E, 2 SC | 6 ano 10 ne |
4 dermis 10 hypodermis 2 muscle |
X=15 | X=42.8 |
Pacienti, kteří zemřeli, jsou uvedeni tučně.
AS léčba, léčba angiosarkomu (1, operace ; 2, radioterapie ; 3, chemoterapie ); DoI, hloubka invaze (2, dermis; 3, hypodermis; 4, sval); E, epiteloidní; HP, histopatologický (1, vazoformní ; 2,solidní; 3, smíšený); IDC, invazivní duktální karcinom; ILC, invazivní lobulární karcinom; M, muž; ns, neuvedeno; PR, postradiační angiosarkom; Rx, záření; SC, vřetenobuněčný; ST, Stewartův-Trevesův angiosarkom; W, žena; WJ, Wilsonův-Jonesův angiosarkom: X, průměr.
Srovnání proměnných mezi přeživšími a pacienty, kteří zemřeli na kožní angiosarkom.
| Proměnná | Žijící (n=6) | Zemřelí (n=10) | |
|---|---|---|---|
| Věk, průměr, y | 61 | 71 | |
| Ženy | 3 | 8 | |
| Muži | 3 | 2 | |
| Postradiační | 4 | 6 | |
| Idiopatický | 2 | 3 | |
| Lymfedém-přidružený | 0 | 1 | |
| Trup | 4 | 6 | |
| Hlava a krk | 2 | 3 | |
| Horní končetiny | 0 | 1 | |
| Velikost, cm | 3.6 | 13,1 | |
| Doba od radioterapie, mo | 82,25 | 110.6 | |
| Dávka záření, Gy | 44 | 47 | |
| Prs | 3 | 7 | |
| Seminom | 1 | ||
| Chirurgie | 6 | 8 | |
| Radioterapie | 1 | 3 | |
| Chemoterapie | 0 | 8 | |
| Vazoformativa | 2 | 2 | |
| Pevné | 2 | 6 | |
| Smíšené | 2 | 2 | |
| Nekróza | 1 | 5 | |
| Dermis | 2 | 2 | |
| Podkoží | 4 | 6 | |
| Svalovina | 0 | 2 | |
| Mitózy | 10 | 18.3 | |
| Přežití, mo | 81,8 | 19.4 | |
Diskuse
Kutánní angiosarkom je velmi vzácný nádor, o čemž svědčí skutečnost, že se nám podařilo shromáždit údaje pouze o 16 nádorech diagnostikovaných za období 14 let v onkologické nemocnici. Celkově je kožní angiosarkom poněkud častější u starších mužů. Je to proto, že nejčastější formou kožního angiosarkomu v běžné populaci je primární angiosarkom hlavy a krku (známý také jako idiopatický nebo Wilson-Jonesův angiosarkom)(obr. 1), který postihuje spíše starší muže.5,7,17 Postradiační angiosarkom (obr. 2) je nyní druhou nejčastější formou kožního angiosarkomu v důsledku častějšího používání radioterapie namísto radikální mastektomie k léčbě karcinomu prsu.2,13,18 Tato změna vedla také ke snížení frekvence výskytu lymfedémového angiosarkomu, který je v současnosti nejméně častou formou tohoto nádoru. Výskyt různých forem kožního angiosarkomu v našem souboru se neshoduje s údaji v literatuře, protože v naší nemocnici se léčí velké množství žen s karcinomem prsu, což vysvětluje, proč nejčastější formou kožního angiosarkomu v naší nemocnici byl postradiační angiosarkom. Tato převaha případů karcinomu prsu také vysvětluje, proč 11 ze 16 pacientů byly ženy. Byl zaznamenán pouze 1 případ angiosarkomu spojeného s lymfedémem (obr. 3), což se však shoduje s mírou prevalence uváděnou jinde. Pacientka měla chronický lymfedém levé paže sekundárně po disekci axilárních lymfatických uzlin provedené v rámci léčby karcinomu prsu před 22 lety.
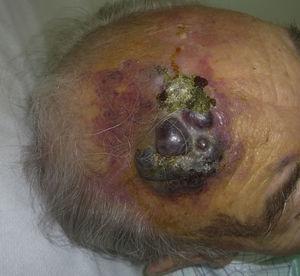
Červenavě fialový plak s nodulárními oblastmi na čele staršího muže.
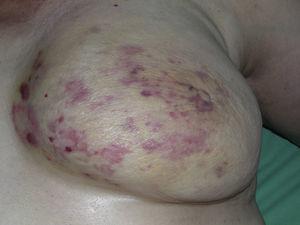
Mnohočetné makuly a erytematózní skvrny s několika načervenalými papulami na nažloutlé kůži na prsu ozářeném v důsledku rakoviny.
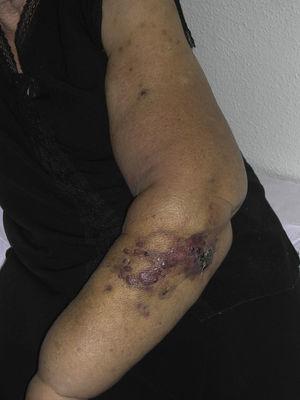
Červenavě fialové papuly a noduly s oblastmi podobnými modřinám na paži s lymfedémem sekundárně po operaci karcinomu prsu
S výjimkou 1 ženy, která měla idiopatický nebo Wilson-Jonesův angiosarkom, měly ostatní ženy v našem souboru (n=10) v anamnéze karcinom prsu. U devíti z nich se jednalo o invazivní duktální karcinomy. Vzhledem k tomu, že 80 % karcinomů prsu v běžné populaci tvoří invazivní duktální karcinomy a pouze 10 % lobulární karcinomy, je vysoká prevalence invazivního duktálního karcinomu v naší studii pravděpodobně odrazem vysoké prevalence tohoto karcinomu v běžné populaci, nikoli zvláštní souvislosti mezi postradiačním kožním angiosarkomem a invazivním duktálním karcinomem, jak by se mohlo zdát z našich výsledků. Jinými slovy, podíly pozorované v našem souboru jsou v souladu s podíly popsanými u různých karcinomů prsu v obecné populaci. V naší studii byly původně zahrnuty 2 případy angiosarkomu prsu, které stojí za zmínku. Uvádí se, že angiosarkom prsu má tendenci být kožní, pokud je vyvolán radioterapií, a parenchymový, pokud není.19 V našem souboru byly z 18 původně identifikovaných případů kožního angiosarkomu 2 případy angiosarkomu prsu, které nebyly vyvolány radioterapií. Při přezkoumání těchto případů jsme zjistili, že primární lokalizací nádoru byl parenchym mléčné žlázy, nikoli kůže. Tyto nádory byly proto ze studie vyřazeny, protože se jednalo o sekundární, nikoli primární nádory. Ve všech zbývajících případech se jednalo o primární angiosarkom prsu indukovaný radiací, což je v souladu se zprávami v literatuře.
Nejčastější lokalizací angiosarkomu v našem souboru byl prs (10 případů), nikoliv obličej nebo kůže hlavy, jak by se dalo očekávat. Jediný případ, který se týkal končetin, byl angiosarkom spojený s lymfedémem. Skutečnost, že nejčastějším místem výskytu nádoru byl prs, by se opět dala vysvětlit převahou nádorů vyvolaných zářením v našem souboru.
Ačkoli je doba latence mezi vystavením radioterapii a vznikem angiosarkomu podle zpráv v literatuře velmi variabilní (3-50 let), bývá delší (v průměru 25 let), pokud je onemocnění léčené zářením benigní. Doba latence uváděná u maligních onemocnění je kratší (přibližně 10-15 let), s výjimkou angiosarkomu prsu, u něhož byla popsána průměrná doba přibližně 5 let12. Důvod této kratší doby v případě angiosarkomu prsu je nejasný, i když bylo navrženo několik teorií, včetně velkého objemu ozářené kůže, přítomnosti přidruženého lymfedému, faktorů, které jsou pravděpodobně vlastní prsu, a možného synergického účinku s chemoterapií.13,14 V našich 10 případech po ozáření byl angiosarkom diagnostikován v průměru po 8,2 letech a v 90 % případů uplynulo nejméně 5 let. U angiosarkomu nebyla zaznamenána žádná souvislost mezi dobou latence a prognózou. V našem souboru byla průměrná doba od radioterapie do vzniku angiosarkomu poněkud delší u pacientů, kteří zemřeli (110,6 měsíce), než u těch, kteří přežili (82,25 měsíce). V jediném případě Stewartova-Trevesova syndromu podstoupil pacient před 22 lety disekci lymfatických uzlin. Doby latence uváděné u angiosarkomu spojeného s lymfedémem jsou velmi variabilní a pohybují se v rozmezí od 1 do 30 let a v průměru 10 let. Stewartův-Trevesův syndrom představuje 90 % všech případů angiosarkomu spojeného s lymfedémem.9,11 Angiosarkom však může vzniknout i u jiných forem lymfedému, jako je vrozený lymfedém, filární lymfedém a lymfedém sekundárně po disekci lymfatických uzlin v jiných částech těla.10
Velikost nádoru je v současnosti nejrozšířenějším prognostickým ukazatelem kožního angiosarkomu a často se uvádí, že angiosarkomy o velikosti 5 cm a více mají horší prognózu než menší nádory.5,17 V našem souboru jsme pozorovali rozdíly v průměrné velikosti nádoru mezi přeživšími a nepřeživšími pacienty. Ti, kteří přežili, měli průměrnou velikost nádoru 3,6 cm, což bylo více než třikrát méně než průměrná velikost (13,1 cm) ve skupině pacientů, kteří zemřeli. Naše výsledky tak podporují přesvědčení, že větší nádory jsou u angiosarkomu spojeny s horšími výsledky,a zdůrazňují důležitost diagnostiky nádoru, když začíná jako léze podobná modřině, ačkoli to je obzvláště obtížné u angiosarkomů vlasové části hlavy a ještě více, když má pacient stále vlasy (obr. 4). Postradiační angiosarkom prsu se diagnostikuje snadněji, protože každá perzistující léze v této oblasti s vaskulárním nebo modřinovitým vzhledem by měla být bioptována. Jedním z potenciálně užitečných diagnostických vodítek je nažloutlé halo (odpovídající hemosiderinu) kolem podezřelé léze (obr. 5), protože tento znak jsme u benigních cévních proliferací na ozářené kůži nikdy nepozorovali. Postradiační angiosarkom a atypické cévní proliferace na ozářené kůži je třeba odlišit histologicky, ale pomoci mohou klinické znaky. Atypické cévní proliferace bývají mnohem menší než angiosarkomy a mají také obecně kratší dobu latence.20 Na rozdíl od angiosarkomů jsou tyto proliferace omezeny na povrchovou a střední dermis a nezasahují do podkoží. Kromě toho histologie nevykazuje charakteristické jaderné atypie pozorované u angiosarkomu, ani mnohočetné vrstvy endoteliálních buněk nebo mitotické figury. Přesto může být někdy obtížné tyto dvě jednotky odlišit a existují zprávy o koexistenci těchto dvou lézí ve stejném ozářeném prsu. Byly dokonce zaznamenány případy radiací indukovaných atypických vaskulárních proliferací přecházejících v angiosarkom.21 Ve velmi komplikovaných případech může být užitečné pátrat po nadměrné expresi genu MYC, pokud se provádí imunohistochemické vyšetření, nebo po amplifikaci, pokud se použije fluorescenční in situ hybridizace. Amplifikace genu MYC je poměrně častá u sekundárních angiosarkomů, ale nezdá se, že by se vyskytovala u atypických cévních proliferací na ozářené kůži.22
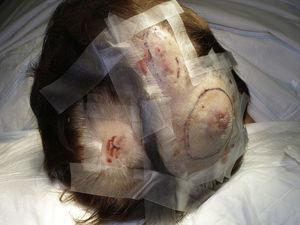
Multifokální erytematózní uzlíky na kůži hlavy.
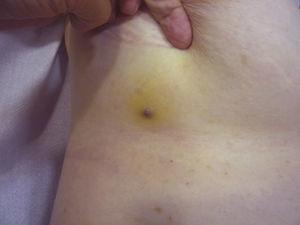
Červenavě fialová papula o velikosti menší než 1 cm obklopená nažloutlou oblastí kůže.
Druhým nejrozšířenějším prognostickým markerem u angiosarkomu je věk. V našem souboru byli pacienti, kteří přežili, v průměru mladší než ti, kteří zemřeli (62 versus 71 let). Bez ohledu na to 10 z 16 pacientů v našem souboru zemřelo do konce studie, což potvrzuje špatnou prognózu obecně spojovanou s angiosarkomem.
Na rozdíl od jiných sarkomů nepovažuje American Joint Committee on Cancer histologický grade za relevantní prognostický marker u angiosarkomu. Několik autorů však naznačilo, že určité histopatologické znaky mohou mít vliv na prognózu. Některé nedávné studie například tvrdí, že převažující solidní vzorec může být u angiosarkomu hlavy a krku relativně dobrým prognostickým markerem.3,16 V našem souboru tomu tak nebylo, protože 6 z 10 pacientů, kteří zemřeli, mělo tento vzorec ve srovnání s pouhými 2 ze 6 pacientů, kteří na konci studie stále žili (obr. 6 a 7).
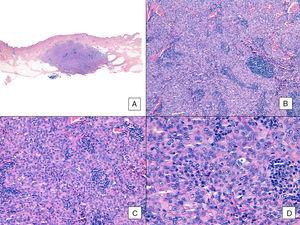
Angiosarkom s převažujícím solidním vzorem. A, Panoramatický snímek ukazuje invazi do střední a hluboké retikulární dermis a hypodermis (hematoxylin-eozin, původní zvětšení ×10). B, Hustě buněčný nádor, který zničil již existující struktury a je doprovázen nodulárními lymfoidními infiltráty (hematoxylin-eozin, původní zvětšení ×100). C a D, Detailní pohled ukazující převahu epiteloidních buněk doprovázených lymfocytárním infiltrátem (hematoxylin-eozin ×200 a ×400).
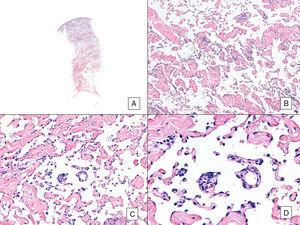
Angiosarkom s převažujícím vazoformním obrazem. A, Panoramatický snímek zobrazující infiltraci dermis až do hypodermis (hematoxylin-eosin, původní zvětšení ×10). B, Neoplastické cévní prostory s výraznou disekcí kolagenu (hematoxylin-eozin, původní zvětšení ×100). C a D, Neoplastické cévy disekované neoplastickými endoteliálními buňkami, které zůstávají „plovoucí“ v dermis (znak promontoria) (hematoxylin-eosin, původní zvětšení ×200 a ×400).
Další potenciálně rizikové histologické faktory navrhované v literatuře jsou přítomnost nekrózy, epiteloidní morfologie a větší hloubka invaze.5,7 Naše výsledky tyto potenciální markery podporují, protože všechny 3 proměnné byly častější ve skupině nepřeživších. Nekróza byla pozorována u 5 z 10 pacientů, kteří zemřeli, a pouze u 1 ze 6 pacientů, kteří přežili. Stejně tak pouze 2 pacienti s převažujícím vřetenobuněčným obrazem byli na konci studie stále naživu. Naopak žádný z pacientů, kteří zemřeli, neměl tento vzorec. Nakonec zemřeli pouze 2 pacienti s postižením svalových rovin. Průměrný počet mitóz byl také vyšší ve skupině nepřeživších (18,3 vs 10).
Léčbou volby kožního angiosarkomu je chirurgická excize s širokými okraji následovaná radioterapií.6 Častá multifokální povaha angiosarkomu a jeho sklon k subklinickému šíření často komplikuje dosažení jasných okrajů. Navíc neexistuje všeobecná shoda na tom, co představuje adekvátní okraj, a většina studií uvádí nepřesné informace, například excizi s „širokými okraji“. V našem souboru byl chirurgický zákrok metodou volby u 14 ze 16 pacientů. Tam, kde to bylo technicky možné, byly použity okraje o délce 3 cm, v ostatních případech byl učiněn pokus o vyčištění pomocí okrajů o délce 2 cm. U 14 pacientů byla provedena radikální operace. Další 2 pacienti nebyli považováni za kandidáty na operaci a podstoupili paliativní chemoterapii. Během sledování zemřeli. Adjuvantní radioterapie chirurgického ložiska byla použita pouze u 4 pacientů. Toto nízké využití radioterapie v léčbě angiosarkomu v našem souboru lze pravděpodobně vysvětlit velkým počtem případů postradiačních angiosarkomů v našem souboru. Jinými slovy, pravděpodobně to bylo ovlivněno určitou mírou rezistence k použití záření v těchto případech. Použití radioterapie u postradiačního angiosarkomu však nachází oporu v literatuře a někdy se dokonce podává jako monoterapie, bez chirurgického zákroku.23,24 Chemoterapie byla použita u poloviny pacientů. Jak již bylo uvedeno, měla pouze paliativní úlohu a ve všech případech byla spojena se špatným výsledkem. Ačkoli monoterapie paklitaxelem nebyla v našem souboru nejčastěji používaným chemoterapeutickým režimem (protože případy byly diagnostikovány před nějakou dobou), tento režim se nyní používá jako možnost první volby ve většině případů. Navzdory původně vyvolaným očekáváním se angiogenní léky (sunitinib, sorafenib, bevacizumab, thalidomid) v tomto případě nepoužívají vzhledem k jejich neuspokojivým výsledkům.
Přes malý počet případů v naší sérii kožního angiosarkomu, který nám bránil v provedení statistických analýz, jsme pozorovali, že větší velikost nádoru a vyšší věk byly spojeny s horší prognózou. Méně zřetelná souvislost byla také pozorována mezi špatnou prognózou a následujícími histologickými znaky: přítomností nekrózy, převahou epiteloidních buněk, invazí do hlubších vrstev a větším počtem mitóz.
Etická prohlášeníOchrana lidí a zvířat
Autoři prohlašují, že pro účely této studie nebyly prováděny žádné testy na lidech ani zvířatech.
Důvěrnost údajů
Autoři prohlašují, že se řídili protokolem své nemocnice o zveřejňování údajů týkajících se pacientů.
Právo na soukromí a informovaný souhlas
Autoři prohlašují, že se v tomto článku neobjevují žádné soukromé údaje pacientů.
Konflikty zájmů
Autoři prohlašují, že nejsou ve střetu zájmů.
Podle prohlášení autorů se v tomto článku neobjevují žádné soukromé údaje.